
11.10: Hydroboration–Oxidation of Alkynes
- Page ID
- 28239

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Hydroboration Reactions
Diborane reacts readily with alkynes, but the formation of substituted alkene products leaves open the possibility of a second addition reaction. A clever technique for avoiding this event takes advantage of the fact that alkynes do not generally suffer from steric hindrance near the triple-bond (the configuration of this functional group is linear). Consequently, large or bulky electrophilic reagents add easily to the triple-bond, but the resulting alkene is necessarily more crowded or sterically hindered and resists further additions. The bulky hydroboration reagent needed for this strategy is prepared by reaction of diborane with 2-methyl-2-butene, a highly branched alkene. Because of the alkyl branching, only two alkenes add to a BH3 moiety (steric hindrance again), leaving one B-H covalent bond available for reaction with an alkyne, as shown below. The resulting dialkyl borane is called disiamylborane, a contraction of di-secondary-isoamylborane (amyl is an old name for pentyl).
An important application of disiamylborane is its addition reaction to terminal alkynes. As with alkenes, the B-H reagent group adds in an apparently anti-Markovnikov manner, due to the fact that the boron is the electrophile, not the hydrogen. Further addition to the resulting boron-substituted alkene does not occur, and the usual oxidative removal of boron by alkaline hydrogen peroxide gives an enol which rapidly rearranges to the aldehyde tautomer. Thus, by the proper choice of reagents, terminal alkynes may be converted either to methyl ketones (mercuric ion catalyzed hydration) or aldehydes (hydroboration followed by oxidation).
Hydroboration of internal alkynes is not a particularly useful procedure because a mixture of products will often be obtained, unless the triple-bond is symmetrically substituted. Mercuric ion catalyzed hydration gives similar results.
Hydroboration-Oxidation is a two step pathway used to produce alcohols. The reaction proceeds in an Anti-Markovnikov manner, where the hydrogen (from BH3 or BHR2) attaches to the more substituted carbon and the boron attaches to the least substituted carbon in the alkene bouble bond. Furthermore, the borane acts as a lewis acid by accepting two electrons in its empty p orbital from an alkene that is electron rich. This process allows boron to have an electron octet. A very interesting characteristic of this process is that it does not require any activation by a catalyst. The Hydroboration mechanism has the elements of both hydrogenation and electrophilic addition and it is a stereospecific (syn addition), meaning that the hydroboration takes place on the same face of the double bond, this leads cis stereochemistry.
The Mechanism
Step #1
- Part #1: Hydroboration of the alkene. In this first step the addittion of the borane to the alkene is initiated and prceeds as a concerted reaction because bond breaking and bond formation occurs at the same time. This part consists of the vacant 2p orbital of the boron electrophile pairing with the electron pair of the ? bondof the nucleophile.
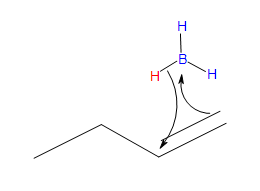
Transition state
.bmp?revision=1&size=bestfit&width=441&height=242)
* Note that a carbocation is not formed. Therefore, no rearrangement takes place.
- Part #2: The Anti Markovnikov addition of Boron. The boron adds to the less substituted carbon of the alkene, which then places the hydrogen on the more substituted carbon. Both, the boron and the hydrogen add simultaneously on the same face of the double bond (syn addition).
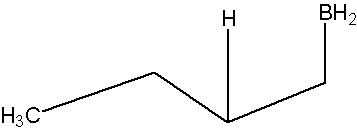
Oxidation of the Trialkylborane by Hydrogen Peroxide
Step #2
- Part #1: the first part of this mechanism deals with the donation of a pair of electrons from the hydrogen peroxide ion. the hydrogen peroxide is the nucleophile in this reaction because it is the electron donor to the newly formed trialkylborane that resulted from hydroboration.

.bmp?revision=1&size=bestfit&width=696&height=239)
- Part 2: In this second part of the mechanism, a rearrangement of an R group with its pair of bonding electrons to an adjacent oxygen results in the removal of a hydroxide ion.
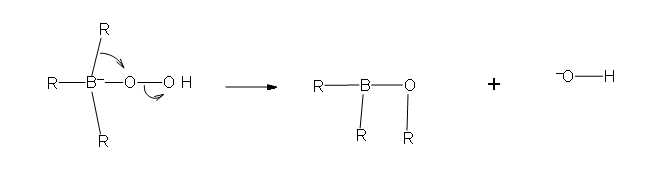
Two more of these reactions with hydroperoxide will occur in order give a trialkylborate
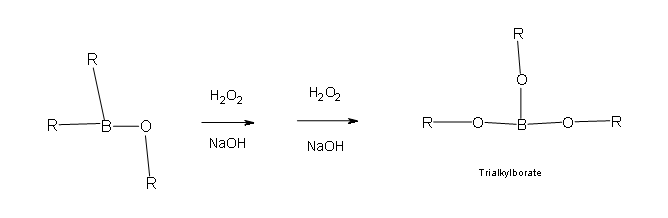
- Part 3: This is the final part of the Oxidation process. In this part the trialkylborate reacts with aqueous NaOH to give the alcohol and sodium borate.

If you need additional visuals to aid you in understanding the mechanism, click on the outside links provided here that will take you to other pages and media that are very helpful as well.
Stereospecific Hydroboration Oxidation of Alkynes
Step 1: Hydroboration of terminal alkynes reacts in an anti-Markovnikov fashion in which the Boron attacks the less substituted carbon which is the least hindered. It is a stereospecific reaction where syn addition is observed as the hydroboration occurs on the same side of the alkyne and results in cis stereochemistry. However, a bulky borane reagent needs to be used to stop at the alkenyl-borane stage. Otherwise, a second hydroboration will occur. In this example, diisoamyl borane or HBsia2, a fairly large and sterically-hindered borane reagent, is used. Both of the alkyne's pi bonds will undergo hydroboration if BH3 (borane) is used by itself.
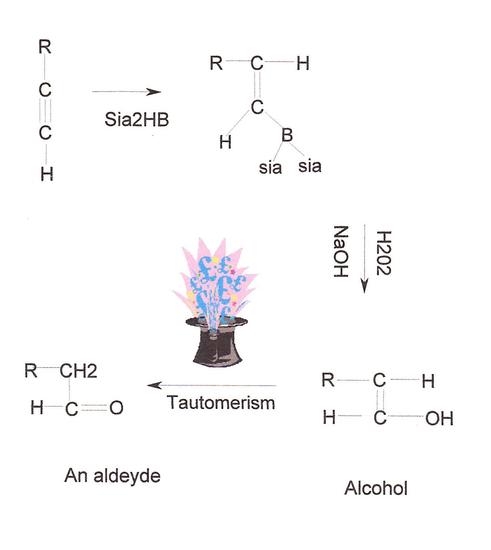
Step 2: Oxidation is the next step that occurs. The resulting alkylborane is oxidized to a vinyl alcohol due to the reaction with a hydroxide in a basic solution such as aqueous sodium hydroxide. A vinyl alcohol is an alcohol that has both an alkene and an -OH group. After the vinyl alcohol is formed, tautomerization takes place. Tautomerism is the interconversion of isomeric compounds due to the migration of a proton. The alcohol, which in this case is a terminal enol, spontaneously and rapidly rearranges due to tautomerism to become an aldehyde since the aldehyde form is much more stable than the enol.
For additional information on hydroboration oxidation: Hydroboration Oxidation
Contributors
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
- Gilbert Torres (UCD)
- Ali Alvandi