
2.4: Protein Folding and Prions
- Page ID
- 165267
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Protein Folding
Proteins are folded and held together by several forms of molecular interactions. The molecular interactions include the thermodynamic stability of the complex, the hydrophobic interactions and the disulfide bonds formed in the proteins.
The biggest factor in a proteins ability to fold is the thermodynamics of the structure. The interaction scheme includes the short-range propensity to form extended conformations, residue-dependent long-range contact potentials, and orientation-dependent hydrogen bonds. The thermodynamics are a main stabilizing force within a protein because if it is not in the lowest energy conformation it will continue to move and adjust until it finds its most stable state. The use of energy diagrams and maps are key in finding out when the protein is in the most stable form possible.
The next type of interaction in protein folding is the hydrophobic interactions within the protein. The framework model and the hydrophobic collapse model represent two canonical descriptions of the protein folding process. The first places primary reliance on the short-range interactions of secondary structure and the second assigns greater importance to the long-range interactions of tertiary structure.6 These hydrophobic interactions have an impact not just on the primary structure but then lead to changes seen in the secondary and tertiary structure as well. Globular proteins acquire distinct compact native conformations in water as a result of the hydrophobic effect.7 When a protein has been folded in the correct way, it usually exists with the hydrophobic core as a result of being hydrated by waters in the system around it which is important because it creates a charged core to the protein and can lead to the creation of channels within the protein. The hydrophobic interactions are found to affect time correlation functions in the vicinity of the native state even though they have no impact on same time characteristics of the structure fluctuations around the native state.7 The hydrophobic interactions are shown to have an impact on the protein even after it has found the most stable conformation in how the proteins can interact with each other as well as folding themselves.
Another type of interaction seen when the protein is folding is the disulfide linkages that form in the protein. (See figure 2.4.2). The disulfide bond, a sulfur- sulfur chemical bond that results from an oxidative process that links nonadjacent (in most cases) cysteine’s of a protein.9 These are a major way that proteins get into their folded form. The types of disulfide bonds are cysteine-cysteine linkage is a stable part of their final folded structure and those in which pairs of cysteines alternate between the reduced and oxidized states.9 The more common is the linkages that cause the protein to fold together and link back on itself compared to the cysteines that are changing oxidation states because the bonds between cysteines once created are fairly stable. The exchange of disulfide bonding targets is catalyzed by protein disulfide isomerase (PDI).
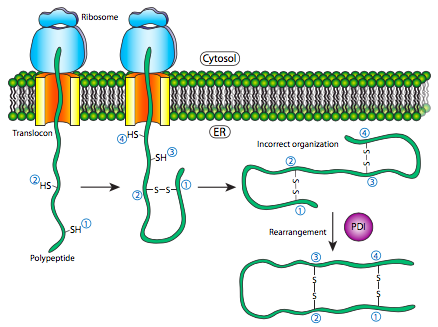
Figure 2.4.2: Disulfide linkages
Denaturation
For proteins, function is dependent on precise structure. Loss of the precise, folded structure of a protein is known as denaturation and is usually accompanied by loss of function. Anyone who has ever worked to purify an enzyme knows how easy it is for one to lose its activity. A few enzymes, such as ribonuclease, are remarkably stable under even very harsh conditions. For most others, a small temperature or pH change can drastically affect activity. The reasons for these differences vary, but relate to 1) the strength of the forces holding the structure together and 2) the ability of a protein to refold itself after being denatured. Let us consider these separately below.

Figure 3.2.18: Denaturation and renaturation of ribonuclease.
Forces Stabilizing Structures
Amino acids are linked one to the other by peptide bonds. These covalent bonds are extraordinarily stable at neutral pHs, but can be broken by hydrolysis with heat under acidic conditions. Peptide bonds, however, only stabilize primary structure and, in fact, are the only relevant force responsible for it. Secondary structure, on the other hand, is generally stabilized by weaker forces, including hydrogen bonds. Hydrogen bonds are readily disrupted by heat, urea, or guanidinium chloride.
Forces stabilizing tertiary structure include ionic interactions, disulfide bonds, hydrophobic interactions, metallic bonds, and hydrogen
bonds. Of these, the ionic interactions are most sensitive to pH changes. Hydrophobic bonds are most sensitive to detergents. Thus,
washing one’s hands helps to kill bacteria by denaturing critical proteins they need to survive. Metallic bonds are sensitive to oxidation/reduction. Breaking disulfide bonds requires either a strong oxidizing agent, such as performic acid or a strong reducing agent
on another disulfide, such as mercaptoethanol or dithiothreitol.
Quaternary structures are stabilized by the same forces as tertiary structure and have the same sensitivities.
Refolding Denatured Proteins
All of the information for protein folding is contained in the primary structure of the protein. It may seem curious then that most proteins do not refold into their proper, fully active form after they have been denatured and the denaturant is removed. A few do, in fact, refold correctly under these circumstances. A good example is bovine ribonuclease (also called RNase). Its catalytic activity is very resistant to heat and urea. However, if one treats the enzyme with mercaptoethanol (which breaks disulfide bonds) prior to urea treatment and
heating, activity is lost, indicating that the covalent disulfide bonds help stabilize the overall enzyme structure. If one allows the enzyme
mixture to cool back down to room temperature, over time some enzyme activity reappears, indicating that ribonuclease can re-fold under the proper conditions.
Irreversible Denaturation
Most enzymes, however, do not behave like ribonuclease. Once denatured, their activity cannot be recovered to any significant extent. This may seem to contradict the idea that folding information is inherent to the sequence of amino acids in the protein. It does not. The reason most enzymes can’t refold properly is due to two phenomena. First, normal folding may occur as proteins are being made. Interactions among amino acids early in the synthesis are not “confused" by interactions with amino acids later in the synthesis because those amino acids aren’t present as protein synthesis starts. In many cases, the proper folding of newly made polypeptides is also assisted by special proteins called chaperones. Chaperones bind to newly made proteins, preventing interactions that might result in misfolding. Thus, early folding and the assistance of chaperones eliminate some potential “wrong-folding" interactions that can occur if the entire sequence was present when folding started.
Denatured full-length polypeptides have many more potential wrong folds that can occur. A second reason most proteins don’t refold properly after denaturation is probably that folding, like any other natural phenomenon, is driven by energy minimization. Though the folded structure may have a low energy, the path leading to it may not be all downhill. Like a chemical reaction that has energies of activation that must be overcome for the reaction to occur, folding likely has peaks and valleys of energy that do not automatically lead directly to the proper fold. Again, folding during synthesis leads the protein along a better-defined path through the energy maze of folding that denatured full-length proteins can’t navigate.
Misfunctions
Proteins can malfunction for several reasons. When a protein is miss folded it can lead to denaturation of the protein. Denaturation is the loss of protein structure and function.1 The miss folding does not always lead to complete lack of function but only partial loss of functionality. The miss functioning of proteins can sometimes lead to diseases in the human body.
Alzheimer's Disease
Alzheimer's Disease (AD) is a neurological degenerative disease that affects around 5 million Americans, including nearly half of those who are age 85 or older.10 The predominant risk factors of AD are age, family history, and heredity. Alzheimer’s disease typically results in memory loss, confusion of time and place, misplacing places, and changes in mood and behavior.11 AD results in dense plaques in the brain that are comprised of fibrillar β-amyloid proteins with a well-orders β-sheet secondary structure.12 These plaques visually look like voids in the brain matter (see figure 2.4.3) and are directly connected to the deterioration of thought processes. It has been determined that AD is a protein misfolding disease, where the misfolded protein is directly related to the formation of these plaques in the brain.13 It is yet to be fully understood what exactly causes this protein misfolding to begin, but several theories point to oxidative stress in the brain to be the initiating factor. This oxidation results in damage to the phospholipids in the brain, which has been found to result in a faster accumulation of amyloid β-proteins.

Figure 2.4.3: The figure showing combination of two brain diagrams in one for comparison. In the left normal brain, in the right brain of a person with Alzheimer’s disease.
Cystic Fibrosis
Cystic Fibrosis (CF) is a chronic disease that affects 30,000 Americans. The typical affects of CF is a production of thick, sticky mucus that clogs the lungs and leads to life-threatening lung infection, and obstructs the pancreas preventing proper food processing.15 CF is caused by protein misfolding. This misfolding then results in some change in the protein known as cystic fibrosis transmembrane conductance regulator (CFTR), which can result in this potentially fatal disease.16 In approximately 70% of CF cases, a deletion of phenylalanine at position 508 in the CFTR is deleted. This deletion of Phe508 seems to be directly connected to the formation of CF.17 The protein misfolding that results in CF occurs prior to birth, but it is not entirely clear as to why.
Resources
- Garrett, R.H., and Grisham, C.M. Biochemistry fourth edition; Brooks/Cole. Australia, 2010. p. 93-95, 135, 143, 160.
- Chang, D.T., Syu Y., Lin, P., Predicting the protein-protein interactions using primary structures with predicted protein surface. BCM Bioinformatics. Jan 2010
- Bodis, P., Schwartz, E., Koepf, M. Cornelissen, J. Rowan A.E., Roeland J. Nolte, M. and Woutersen S. Vibrational self-trapping in beta-sheet structures observed with femtosecond nonlinear infrared spectroscopy. The Journal Of Chemical Physics 131, 2009
- Yasuda, S., Yoshidome, T. Oshima, H. Kodama R., Harano, Y. and Kinoshita, M. Effects of side-chain packing on the formation of secondary structures in protein folding. The Journal of Chemical Physics 132, 2010
- Papandreoul, N., Berezovsky I.N., Lopes, A., Eliopoulos, E., and Chomilier, J. Universal positions in globular proteins from observation to simulation. Eur. J. Biochem. 271, 4762–4768 (2004)
- Hausratha, A.C. A Kinetic theory of tertiary contact formation coupled to the helix-coil transition in polypeptides. The Journal of Chemical Physics. 2006
- Cieplaka M. and Niewieczerza S. Hydrodynamic interactions in protein folding. Journal of Chemical Physics. 130 2009
- Gront, D. Kolinskia, A. and Skolnick, J. A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics. Journal of Chemical Physics. Vol. 115 2001
- Kadokura, H., Katzen F. and Beckwith, J. Protein disulfide bond formation in prokatyotes. Annual Review Biochem 2003.
- Alzheimer's Disease. Centers for Disease Control and Prevention. 2010, http://www.cdc.gov/aging/aginginfo/alzheimers.htm.
- Alzheimer's Association. 2011, http://www.alz.org.
- Riordan, J.M. et al. Identification of the cysticfibrosisgene: cloning and characterization of complementary DNA. Science 1989, 245, 1066-1073.
Prions
A prion is an infectious agent composed of protein in a misfolded form. This is the central idea of the Prion Hypothesis, which remains debated. This is in contrast to all other known infectious agents (virus /bacteria/fungus/parasite) which must contain nucleic acids (either DNA, RNA, or both). The word prion, coined in 1982 by Stanley B. Prusiner, is derived from the words protein and infection. Prions are responsible for the transmissible spongiform encephalopathies in a variety of mammals, including bovine spongiform encephalopathy (BSE, also known as “mad cow disease”) in cattle and Creutzfeldt–Jakob disease (CJD) in humans. All known prion diseases affect the structure of the brain or other neural tissue, are currently untreatable and universally fatal.

Prions propagate by transmitting a misfolded protein state. When a prion enters a healthy organism, it induces existing, properly folded proteins to convert into the disease-associated prion form; it acts as a template to guide the misfolding of more proteins into prion form. These newly-formed prions can then go on to convert more proteins themselves; triggering a chain reaction. All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly-packed beta sheets. Amyloid aggregates are fibrils, growing at their ends, and replicating when breakage causes two growing ends to become four growing ends.
The incubation period of prion diseases is determined by the exponential growth rate associated with prion replication, which is a balance between the linear growth and the breakage of aggregates. Propagation of the prion depends on the presence of normally-folded protein in which the prion can induce misfolding; animals which do not express the normal form of the prion protein cannot develop nor transmit the disease.
All known mammalian prion diseases are caused by the so-called prion protein, PrP. The endogenous, properly-folded form is denoted PrPC (for Common or Cellular) while the disease-linked, misfolded form is denoted PrPSc (for Scrapie, after one of the diseases first linked to prions and neurodegeneration. ) The precise structure of the prion is not known, though they can be formed by combining PrPC, polyadenylic acid, and lipids in a Protein Misfolding Cyclic Amplification (PMCA) reaction.
Proteins showing prion-type behavior are also found in some fungi, which has been useful in helping to understand mammalian prions. Fungal prions do not appear to cause disease in their hosts.
The first hypothesis that tried to explain how prions replicate in a protein-only manner was the heterodimer model. This model assumed that a single PrPSc molecule binds to a single PrPC molecule and catalyzes its conversion into PrPSc. The two PrPSc molecules then come apart and can go on to convert more PrPC. However, a model of prion replication must explain both how prions propagate, and why their spontaneous appearance is so rare. Manfred Eigen showed that the heterodimer model requires PrPSc to be an extraordinarily effective catalyst, increasing the rate of the conversion reaction by a factor of around 1015. This problem does not arise if PrPSc exists only in aggregated forms such as amyloid, where cooperativity may act as a barrier to spontaneous conversion. What is more, despite considerable effort, infectious monomeric PrPSc has never been isolated.

An alternative model assumes that PrPSc exists only as fibrils, and that fibril ends bind PrPC and convert it into PrPSc. If this were all, then the quantity of prions would increase linearly, forming even longer fibrils. But exponential growth of both PrPSc and of the quantity of infectious particles is observed during prion disease. This can be explained by taking into account fibril breakage. A mathematical solution for the exponential growth rate resulting from the combination of fibril growth and fibril breakage has been found.

The protein-only hypothesis has been criticised by those who feel that the simplest explanation of the evidence to date is viral. For more than a decade, Yale University neuropathologist Laura Manuelidis has been proposing that prion diseases are caused instead by an unidentified slow virus. In January 2007, she and her colleagues published an article reporting to have found a virus in 10%, or less, of their scrapie-infected cells in culture.
The virion hypothesis states that TSEs are caused by a replicable informational molecule (likely to be a nucleic acid) bound to PrP. Many TSEs, including scrapie and BSE, show strains with specific and distinct biological properties, a feature which supporters of the virion hypothesis feel is not explained by prions.
Recent studies propagating TSE infectivity in cell-free reactions and in purified component chemical reactions strongly suggest against TSE’s viral nature. Using a similar defined recipe of multiple components (PrP, POPG lipid, RNA), Jiyan Ma and colleagues generated infectious prions from recombinant PrP expressed from E. coli, casting further doubt on this hypothesis.
Key Points
- Prions are responsible for the transmissible spongiform encephalopathies in a variety of mammals, including bovine spongiform encephalopathy and Creutzfeldt–Jakob disease in humans. All known prion diseases affect the structure of neural tissue, are currently untreatable and universally fatal.
- Prions propagate by transmitting a misfolded protein state. When a prion enters a healthy organism, it induces existing, properly-folded proteins to convert into the disease-associated, prion form; it then acts as a template to guide the misfolding of more proteins into prion form.
- All known mammalian prion diseases are caused by the so-called prion protein, PrP. The endogenous, properly folded, form is denoted PrPC while the disease-linked, misfolded form is denoted PrPSc.The precise structure of the prion is not known.
- The first hypothesis to explain how prions replicate in a protein-only manner was the heterodimer model, which assumed that a single PrPSc molecule binds to a single PrPC molecule and catalyzes its conversion into PrPSc. The two PrPSc molecules then come apart and can go on to convert more PrPC.
- An alternative model assumes that PrPSc exists only as fibrils and that fibril ends bind PrPC and convert it into PrPSc. The exponential growth of both PrPSc and of the quantity of infectious particles observed during prion disease can be explained by taking fibril breakage into account.
- The protein-only hypothesis has been criticised by those who feel that the simplest explanation of the evidence to date is viral. However, recent studies propagating TSE infectivity in cell-free reactions and in purified component chemical reactions strongly suggest against TSE’s viral nature.
Key Terms
- Amyloid: Insoluble fibrous protein aggregates sharing specific structural traits. They arise from at least 18 inappropriately folded versions of proteins and polypeptides present naturally in the body. These misfolded structures alter their proper configuration such that they erroneously interact with one another or other cell components forming insoluble fibrils. They have been associated with the pathology of more than 20 serious human diseases in that, abnormal accumulation of amyloid fibrils in organs may lead to amyloidosis, and may play a role in various neurodegenerative disorders.
- Creutzfeldt–Jakob disease: A degenerative neurological disorder (brain disease) that is incurable and invariably fatal. In CJD, the brain tissue develops holes and takes on a sponge-like texture, due to a type of infectious protein called a prion.
- prion: A self-propagating misfolded conformer of a protein that is responsible for a number of diseases that affect the brain and other neural tissue.
LICENSES AND ATTRIBUTIONS
CC LICENSED CONTENT, SHARED PREVIOUSLY
- Curation and Revision. Provided by: Boundless.com. License: CC BY-SA: Attribution-ShareAlike
CC LICENSED CONTENT, SPECIFIC ATTRIBUTION
- Subviral agent. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Subviral_agent%23Subviral_agents. License: CC BY-SA: Attribution-ShareAlike
- Virology. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Virology. License: CC BY-SA: Attribution-ShareAlike
- Hepatitis delta virus. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Hepatitis_delta_virus%23Virology. License: CC BY-SA: Attribution-ShareAlike
- Virus. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Virus. License: CC BY-SA: Attribution-ShareAlike
- Satellite (biology). Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Satellite_(biology). License: CC BY-SA: Attribution-ShareAlike
- Defective interfering particle. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Defective_interfering_particle. License: CC BY-SA: Attribution-ShareAlike
- General Biology/Classification of Living Things/Viruses. Provided by: Wikibooks. Located at: http://en.wikibooks.org/wiki/General_Biology/Classification_of_Living_Things/Viruses. License: CC BY-SA: Attribution-ShareAlike
- Helper virus. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Helper%20virus. License: CC BY-SA: Attribution-ShareAlike
- Satellite. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Satellite. License: CC BY-SA: Attribution-ShareAlike
- Hepatocellular carcinoma 1. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Hepatocellular_carcinoma_1.jpg. License: Public Domain: No Known Copyright
- Avsunviroidae. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Avsunviroidae. License: CC BY-SA: Attribution-ShareAlike
- Pospiviroidae. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Pospiviroidae. License: CC BY-SA: Attribution-ShareAlike
- Avocado sunblotch viroid. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Avocado_sunblotch_viroid. License: CC BY-SA: Attribution-ShareAlike
- Viroid. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Viroid. License: CC BY-SA: Attribution-ShareAlike
- Virusoid. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Virusoid. License: CC BY-SA: Attribution-ShareAlike
- viroid. Provided by: Wiktionary. Located at: http://en.wiktionary.org/wiki/viroid. License: CC BY-SA: Attribution-ShareAlike
- Virusoid. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Virusoid. License: CC BY-SA: Attribution-ShareAlike
- Hepatocellular carcinoma 1. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Hepatocellular_carcinoma_1.jpg. License: Public Domain: No Known Copyright
- PSTviroid. Provided by: Wikimedia. Located at: http://commons.wikimedia.org/wiki/File:PSTviroid.png?uselang=he. License: Public Domain: No Known Copyright
- Prion. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Prion. License: CC BY-SA: Attribution-ShareAlike
- prion. Provided by: Wiktionary. Located at: http://en.wiktionary.org/wiki/prion. License: CC BY-SA: Attribution-ShareAlike
- Amyloid. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Amyloid. License: CC BY-SA: Attribution-ShareAlike
- Creutzfeldt. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/Creutzfeldt?Jakob+disease. License: CC BY-SA: Attribution-ShareAlike
- Hepatocellular carcinoma 1. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Hepatocellular_carcinoma_1.jpg. License: Public Domain: No Known Copyright
- PSTviroid. Provided by: Wikimedia. Located at: http://commons.wikimedia.org/wiki/File:PSTviroid.png?uselang=he. License: Public Domain: No Known Copyright
- Histology bse. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Histology_bse.jpg. License: Public Domain: No Known Copyright
- Prion propagation. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Prion_propagation.svg. License: CC BY: Attribution
- Prion replication. Provided by: Wikipedia. Located at: http://en.wikipedia.org/wiki/File:Pr...eplication.png. License: CC BY-SA: Attribution-ShareAlike