
28.6: DNA Sequencing
- Page ID
- 429793
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- Describe briefly how DNA sequencing is carried out.
DNA sequencing determines the order of nucleotide bases within a given fragment of DNA. This information can be used to infer the RNA or protein sequence encoded by the gene, from which further inferences may be made about the gene’s function and its relationship to other genes and gene products. DNA sequence information is also useful in studying the regulation of gene expression. If DNA sequencing is applied to the study of many genes, or even a whole genome, it is considered an example of genomics.
While techniques to sequence proteins have been around since the 1950s, techniques to sequence DNA were not developed until the mid-1970s, when two distinct sequencing methods were developed almost simultaneously, one by Walter Gilbert’s group at Harvard University, the other by Frederick Sanger’s group at Cambridge University. However, until the 1990s, the sequencing of DNA was a relatively expensive and long process. Using radiolabeled nucleotides also compounded the problem through safety concerns. With currently-available technology and automated machines, the process is cheaper, safer, and can be completed in a matter of hours. The Sanger sequencing method was used for the human genome sequencing project, which was finished its sequencing phase in 2003, but today both it and the Gilbert method have been largely replaced by better methods.
Restriction Enzymes
To be able to sequence DNA, it is first necessary to cut it into smaller fragments. What is needed is a way to cleave the DNA molecule at a few precisely-located sites so that a small set of homogeneous fragments are produced. To cut DNA at known locations, researchers use restriction endonucleases enzymes that have been purified from various bacterial species, and which can be purchased from various commercial sources. REs occur naturally in bacteria, where they specifically recognize short stretches of nucleotides in DNA and catalyze double-strand breaks at or near the recognition site (also known as a restriction site). These enzymes are usually named after the bacterium from which they were first isolated. For example, EcoRI and EcoRV are both enzymes from E. coli.
Restriction enzymes like EcoRI are frequently called 6-cutters, because they recognize a 6-nucleotide sequence. Assuming a random distribution of A, C, G and Ts in DNA, probability predicts that a recognition site for a 6-cutter should occur about once for every 4096 bp (46) in DNA. Of course, the distribution of nucleotides in DNA is not random, so the actual sizes of DNA fragments produced by EcoRI range from hundreds to many thousands of base pairs, but the mean size is close to 4000 bp. DNA fragments of this length are useful in the lab, since they long enough to contain the coding sequence for proteins and are well-resolved on agarose gels.
EcoRI recognizes the sequence G A A T T C in double stranded DNA. This recognition sequence is a palindrome with a two-fold axis of symmetry, because reading from 5’ to 3’ on either strand of the helix gives the same sequence. The palindromic nature of the restriction site is more obvious in the figure below. The dot in the center of the restriction site denotes the axis of symmetry. EcoRI catalyzes the hydrolysis of the phosphodiester bonds between G and A on both DNA strands. The restriction fragments generated in the reaction have short single-stranded tails at the 5’-ends. These ends are often referred to as “sticky ends,” because of their ability to form hydrogen bonds with complementary DNA sequences.

Reading DNA Sequences
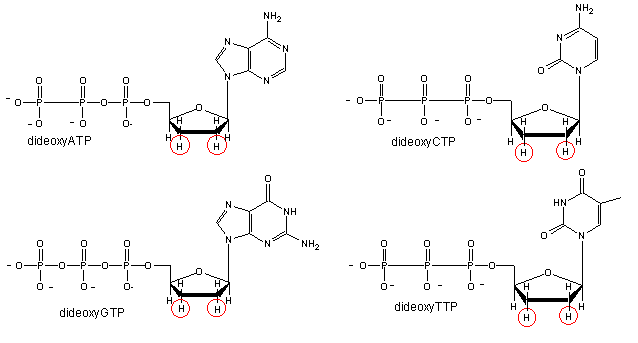
We will discuss one method of reading the sequence of DNA. This method, developed by Sanger won him a second Nobel prize. Sanger sequencing, also known as chain-termination sequencing, requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotidetriphosphates (dNTPs), and modified nucleotides (dideoxyNTPs - ddNTP) that terminate DNA strand elongation. These chain-terminating nucleotides lack a 3′-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a ddNTP is incorporated.
.gif?revision=1)
Four reaction tubes are set up, each containing the template DNA to be sequenced, a primer of known sequence, all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP), and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) which has been fluorescently labeled. Most of the time in a Sanger sequencing reaction, DNA Polymerase will add a proper dNTP to the growing strand it is synthesizing in vitro. But at random locations, it will instead add a ddNTP. When it does, that strand will be terminated at the ddNTP just added. If enough template DNAs are included in the reaction mix, each one will have the labeled ddNTP inserted at a different random location, and there will be at least one DNA terminated at each different nucleotide along its length for as long as the in vitro reaction can take place (about 900 nucleotides under optimal conditions.)
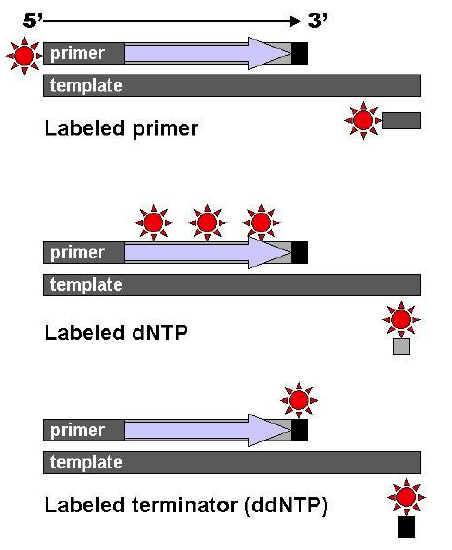
After the reactions are over, the newly synthesized strands can be denatured from the template, and then separated by capillary electrophoresis or other equivalent methods. Since each band differs in length by one nucleotide, and the identity of that nucleotide is known from its fluorescence, the DNA sequence can be read simply from the order of the colors in successive bands.

As each differently-sized fragment exits the capillary column, a laser excites the florescent tag on its terminal nucleotide. From the color of the resulting florescence, a computer can keep track of which nucleotide was present as the terminating nucleotide. The computer also keeps track of the order in which the terminating nucleotides appeared, which is the sequence of the DNA used in the original reaction. In practice, the maximum length of sequence that can be read from a single sequencing reaction is about 700 bp.
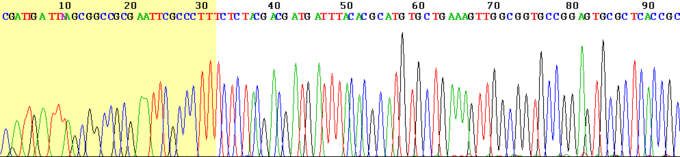
Scientists now know the sequence of all the 3 billion DNA base pairs in the entire human genome. This knowledge was attained by the Human Genome Project (HGP), a $3 billion, international scientific research project that was formally launched in 1990. The project was completed in 2003, two years ahead of its 15-year projected deadline. Determining the sequence of the billions of base pairs that make up human DNA was the main goal of the HGP. Another goal was mapping the location and determining the function of all the genes in the human genome. There are only about 20,500 genes in human beings. If modern methods were used it might bring the cost of sequencing the human genome down from the initial billion dollar range to $100.
You will pretend to sequence a single stranded piece of DNA as shown below. The new nucleotides are added by the enzyme DNA polymerase to the primer, GACT, in the 5' to 3' direction. You will set up 4 reaction tubes, Each tube contains all the dXTP's. In addition, add ddATP to tube 1, ddTTP to tube 2, ddCTP to tube 3, and ddGTP to tube 4. For each separate reaction mixture, determine all the possible sequences made by writing the possible sequences on one of the unfinished complementary sequences below. Cut the completed sequences from the page, determine the size of the polynucleotide sequences made, and place them as they would migrate (based on size) in the appropriate lane of a imaginary gel which you have drawn on a piece of paper. Lane 1 will contain the nucleotides made in tube 1, etc. Then draw lines under the positions of the cutout nucleotides to represent DNA bands in the gel. Read the sequence of the complementary DNA synthesized. Then write the sequence of the ssDNA that was to be sequenced.
- 5' T C A A C G A T C T G A 3' (STAND TO SEQUENCE)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
- 3' G A C T 5' (primer)
Since the DNA fragments have no detectable color, they can not be directly visualized in the gel. Alternative methods are used. In the one described above, radiolabeled ddXTP's where used. Once the sequencing gel is run, it can be dried and the bands visualized by radioautography (also called autoradiography). A place of x-ray film is placed over the dried gel in a dark environment. The radiolabeled bands will emit radiation which will expose the x-ray film directly over the bands. The film can be developed to detect the bands. In a newer technique, the primer can be labeled with a flourescent dye. If a different dye is used for each reaction mixture, all the reaction mixtures can be run in one lane of a gel. (Actually only one reaction mix containing all the ddXTP's together need be performed.) The gel can then be scanned by a laser, which detects fluorescence from the dyes, each at a different wavelength.