
Iron Storage: Ferritin
- Page ID
- 97956

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Iron is necessary for life. It is an essential nutrient and living organisms use it frequently and in large amounts. Iron is necessary in the active sites of enzymes that play roles in respiration, electron transfer reactions, energy metabolism, DNA synthesis, and gene regulation.1 Iron is also necessary for oxygen transport and oxygen storage in the proteins hemoglobin and myoglobin.2
Despite the importance of iron for life, excess iron can be toxic.3 Unregulated free iron can catalyze the production of harmful reactive oxygen species (ROS) and free radicals, most commonly via Fenton chemistry. The Fenton reaction (below) is the reaction between iron(II) ions and hydrogen peroxide to produce the extremely reactive and harmful hydroxyl radical (•OH).1
Fe2+ + H2O2 → Fe3+ + OH- + •OH
ROS are strong oxidizing agents and can cause permanent cell damage, organ failure, and death.4 Furthermore, iron ions have notoriously low solubility. The issues of solubility particularly apply to iron(III) ions which have a solubility of 10-18 M at physiological conditions.1 In environments where oxygen is present, like the bloodstream, iron can form insoluble aggregates, similar to rust.5,6 For example, dying red blood cells produce ~0.54 mmol of iron per day. If that iron is left free in its aerobic environment, it will react with oxygen and form insoluble rust-like compounds. It’s been estimated that a human would have to drink 1013 liters of water every day to avoid “rusty” kidneys.1 Additionally, pathogens need iron to replicate and to survive. Thus, excess iron in an organism could be used both to nourish the organism itself and to nourish infection and disease-causing organisms.4 Due to the biological necessity of iron, the storage and collection of excess iron in a nontoxic form is vital.
The cell’s answer to these obstacles is ferritin. Ferritin is the universal iron storage protein found throughout nature—both in prokaryotic and eukaryotic organisms.4,7 It is found in plants, animals, and bacteria and is necessary for all living things. In fact, deletion of the ferritin gene is lethal.8 Ferritin functions both intracellularly and extracellularly.5 Intracellularly, human ferritin is found in the cytoplasm of the cell.3,4 Extracellularly, human ferritin has been found in serum. It is currently unknown where this extracellular ferritin comes from and how it leaves the cell, but its concentration in blood is used as a common clinical test for iron levels.5 The measurement of serum ferritin is considered as one of the most specific and sensitive blood tests for diagnosing iron deficiency.5
Ferritin solves the previously discussed obstacles of excess iron in the body. Considering the poor solubility of Fe3+ ions, human ferritin is actually rather soluble and gathers excess iron ions without creating insoluble clusters of iron in the body.6 The transport protein transferrin brings excess iron ions to ferritin as Fe2+. The ferritin protein surrounds the collected iron ions to prevent the Fe3+ ions from aggregating in the cytoplasm of the cell or in the bloodstream and to protect them against reaction with any other species present, thus avoiding the production of ROS.6 Any other ligands in the cell that might bind to iron are blocked out and the iron is stored in a stable form.2 Ferritin stores excess iron until iron is needed for one of the many processes in which it plays a part. Iron stored in ferritin can comprise almost 10% of total iron in the body.2
Structurally, ferritin is a hollow cage and the sequestered iron is stored as an iron(III) mineral within the protein shell (Figure 1).2,3
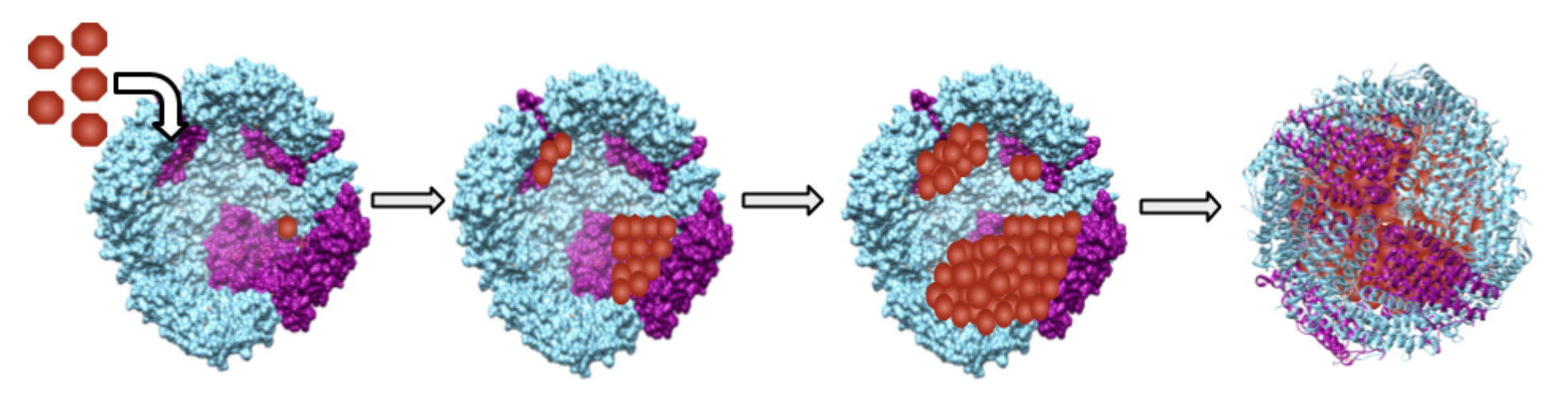
Ferritin is made of subunits. Each subunit has four polypeptide α-helices or spirals.2 Structures of ferritin differ based on the organism which produces it. Most ferritin is made of 24 of these subunits.6 Human ferritin is a 24-subunit, 480 kDa, 12 nm globular protein with a hollow center cavity (cavity diameter of 8 nm).2 One 24-subunit ferritin protein can store up to 4500 iron ions in its hollow center.2 Ferritin is a very good space-saving model for the cell because of its high iron to protein ratio (4500 iron ions/8 nm cavity).2,9 This iron to protein ratio is 200 times that of hemoglobin.9 Up to 24% of ferritin’s weight could be stored iron ions. Meanwhile, bacterial ferritin can be either a 12- or 24-subunit globular protein.6 However, it is thought that the 12-subunit ferritin’s may play an additional role in protecting DNA from oxidative damage, while the 24-subunit bacterial ferritin may control bacterial iron metabolism.3
The subunits of human ferritin are classified as the heavy subunit or “H-chain” and the light subunit or “L-chain” (Figure 2).6 The two subunits, however, are not extremely different in molecular weight or amino acid sequence. The H-chain is made of 178 amino acids with a molecular weight of 21 kDa and the L-chain is made 171 amino acids and weighs 18 kDa.4 The two types of subunits have ~53% protein sequence identity.4 The designations of heavy and light are actually a modern recasting of designations that originally reflected the organs from which the two forms were isolated, “H” for heart and “L” for liver.8
The two types of subunits combine in different ratios to form the 24-subunit protein shell in humans (Figure 2).6 This ratio is dependent on the tissue where the ferritin is synthesized.6 The H subunit is responsible for catalyzing the oxidation of iron(II).8 The L subunit hosts the site of nucleation and storage of iron.8 The ratio of H:L will be higher in tissues where iron oxidation activity is high and iron detoxification is needed, such as the heart or the brain.1,4 Tissues, like those in the spleen, are used more for storage and will have a lower H:L ratio. The human liver produces ferritin that is 50% H and 50% L.
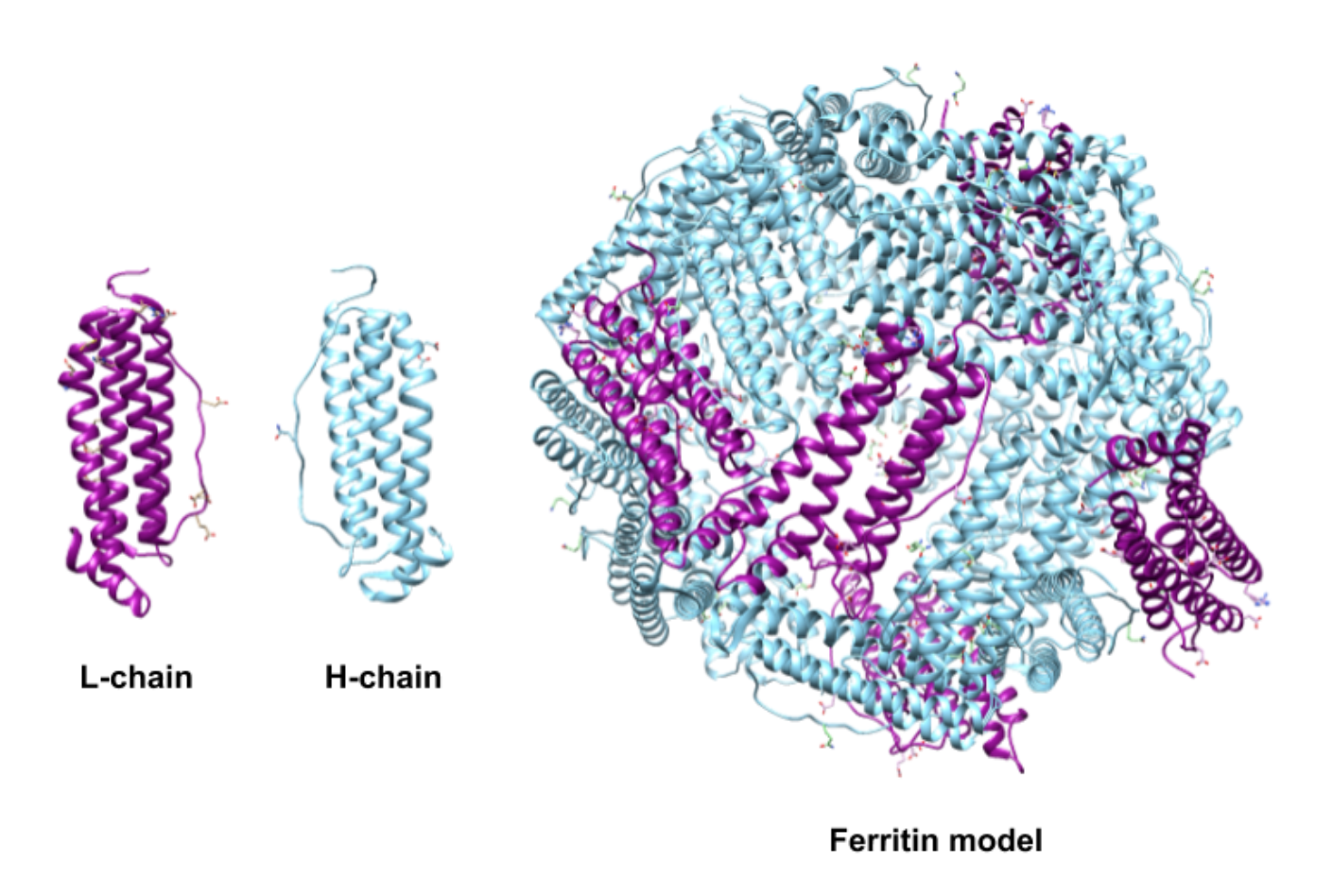
In bacterial ferritin, the H-chain is the only subunit present in the either 12- or 24-subunit shell.2 Amphibious or fish ferritin can be made up of two to three different types of subunits (“H-”,”L-”, and middle “M-chains”).7 The H and M subunits share similar functions of oxidizing iron.12 This report will focus on the H and L subunits of human ferritin.
While iron storage has been determined as an important function, it is unknown if this is the only or even primary function of ferritin.6 Other proposed functions fall mainly under the umbrella of ferritin’s role as an antioxidant. It has been proposed that the main role of ferritin could be in reducing oxidative stress and preventing harmful production of ROS from reactions between iron and oxygen or hydrogen peroxide. The exact mechanism of how ferritin works as an antioxidant is unclear, but its characteristic functions can all contribute.6 For example, the ability to remove O2 or ROS through reaction with Fe2+ and to remove excess Fe2+ from the environment both directly reduce oxidative stress; however, if ferritin is produced by the cell for this purpose has yet to be determined.6
Entrance: Ferritin's Selectivity for Iron
Fe2+ ions are brought to ferritin for storage by the transport protein transferrin. The ions are believed to enter through one of the eight symmetric 3-fold channels or one of the six symmetric 4-fold channels formed by the subunits in the ferritin structure highlighted in Figure 3.6

The 3-fold channels are lined with the polar side chains aspartate and glutamate and make the channel hydrophilic.2 The hydrophilicity of the channel allows for the transport of water, metal cations, and hydrophilic molecules of an appropriate size into and and out of the ferritin center.4 Most studies indicate that the 3-fold channel is the main channel for Fe2+ ions both into and out of the cell.1,3
The 4-fold channels are lined with the non-polar side chain leucine and make the channel hydrophobic. It is widely thought that the 4-fold channels are involved with the diffusion of oxygen and hydrogen peroxide into and out of the ferritin center.1
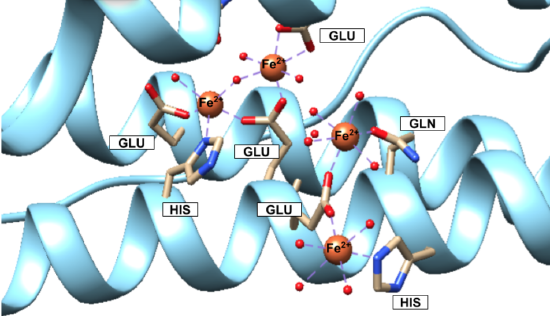
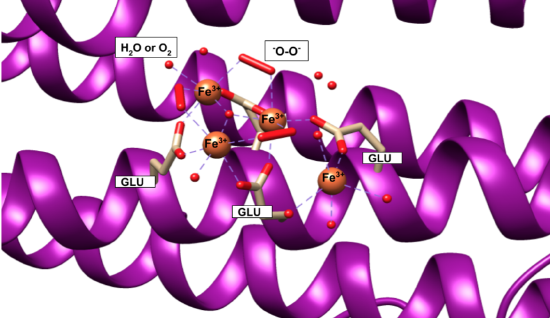
Iron(II) ions are oxidized at the “ferroxidase site” on the H-chain subunits (Figure 4). After conversion to Fe3+, the iron is stored as a crystalline iron/oxy mineral.2 The mineral accumulates on the L-chain (Figure 5).8
 |
 |
|
Figure 4. Human Ferritin H-chain (PDB 4YKH). Fe2+ ions are both shown traveling on their way to the ferroxidase site and at the ferroxidase site, to be covered in detail later. Fe2+ ions can be seen binding to water molecules, oxidizing agents and residues glutamic acid (Glu or GLU), glutamine (Gln or GLN), and histidine (His or HIS). The smaller red balls connecting to the Fe2+ ions via a purple dashed line are the oxidizing agents—oxygen molecules, hydrogen peroxide, or water molecules. |
Figure 5. Human Ferritin L-chain (PDB 5LG8). Fe3+ ions can be seen binding to water and oxygen molecules (small red balls), peroxo groups (long red cylinders), and residues of glutamic acid (Glu or GLU) from the L-chain. Fe3+ is stored as an iron/oxy mineral on the L-chain. This is the beginning of nucleation within the ferritin cavity. |
Figures 4 and 5 show iron(II) at the ferroxidase site and iron/oxy mineral at the nucleation site, according to the available crystal structures. The structure of the entire iron mineral core, however, is actually inconsistent throughout the whole mineral and differs even between identical ferritin shells.
Traditionally, it was believed that the iron(III)/oxy mineral that makes up the core was similar to ferrihydrite and ideally structured as 20% tetrahedral and 80% octahedral; however, as more evidence has become available this model has been replaced by a “polyphasic” or heterogeneous model of the iron core that is thought to be more accurate.2,4 According to the polyphasic model, the mineral structure is heterogeneous in its chemical content and geometric structure.4 X-ray absorption fine structure (EXAFS) studies concluded that the iron core of ferritin was made of iron ions that are surrounded by six or seven oxygen atoms, surrounded by another shell of iron ions.2 This inconsistency leads to the conclusion that the mineral is a heterogeneous, hydrated iron(III)/oxy.3 In other words, the iron core itself is not packed regularly or in a pattern within ferritin.6
The composition of the protein shell does not affect the composition of the iron/oxy mineral, however.6 In fact, two ferritin proteins that are identical in protein makeup (with same H:L ratio) are likely to have a different make-up of their iron cores. The H:L ratio is suspected to affect the geometry of the crystal, due to their inherently different properties. Because the H-chain hosts the ferroxidase site and L-chain hosts the nucleation site, the rate at which the iron is accumulated and the mineral core is formed varies as the ratio does.6 Studies on the crystal structure of the mineral core have found some cores to be more highly ordered and crystalline and other less ordered and more amorphous; these observations are thought to be the result of, or at least correlate to, different H:L ratios.6
Selectivity of iron(II) by ferritin can be explained through hard-soft acid base (HSAB) theory. HSAB theory characterizes metal ions and ligands as either hard, soft, or borderline acids and bases based on the Lewis definition. Characterization, however, should be noted to be a gradient scale for all ions and ligands.HSAB theory states that similarly characterized acids and bases will be more likely to bind with one another; that is, a hard acid will be more likely to bind to a hard base over a soft or borderline base and vice versa. Borderline acids or bases prefer to bind to borderline bases or acids, respectively; however, borderline is more intermediate in character and more flexible in binding. It isn’t uncommon for a borderline acid or base to be more slightly more hard or soft in character.
Ferritin binds an Fe2+ ion for oxidation. Fe2+ is categorized as a borderline acid. The binding sites on the H-chain are glutamic acid (Glu), histidine (His), and glutamine (Gln) residues (Figure 4).6 Glu and Gln are categorized as hard bases, while His is categorized as borderline base. The oxidizing agents—oxygen molecules, hydrogen peroxide, or water molecules all have hard character. Fe2+ as a borderline acid binds well with the binding site of mixed hard and borderline character. HSAB theory supports the selection of Fe2+ as the metal ion for the ligand. The selection of Fe2+ in ferritin is further supported by experimental work where metals like zinc (which also exists in the body) were also shown to have binding abilities to ferritin.5 But, zinc, although it is also borderline, has softer character than Fe2+ and so would not be as stable or favorable with the hard Glu and Gln ligands.10
Because of the initial selection of Fe2+ by the H-chain and the crystalline nature of the iron(III) core, the binding of Fe3+ by the protein is not as key. However, Fe3+ is characterized as a hard acid and the Glu residues on the L-chain and the oxide species in the mineral are also hard, so binding remains preferable.
Selectivity of Fe2+ is further supported by the chelate effect. The chelate effect is an entropic effect where a ligand has multiple donor groups for a metal ion. The entropy increases favorably when a chelator is used to bind because there is one molecule binding to the metal ion through multiple donor atoms, rather than multiple individual ligands. The Fe2+ binding site of ferritin is a chelator. At the ferroxidase site on the H-chain subunit, the binding site of the Fe2+ ions are shown in Figure 6.
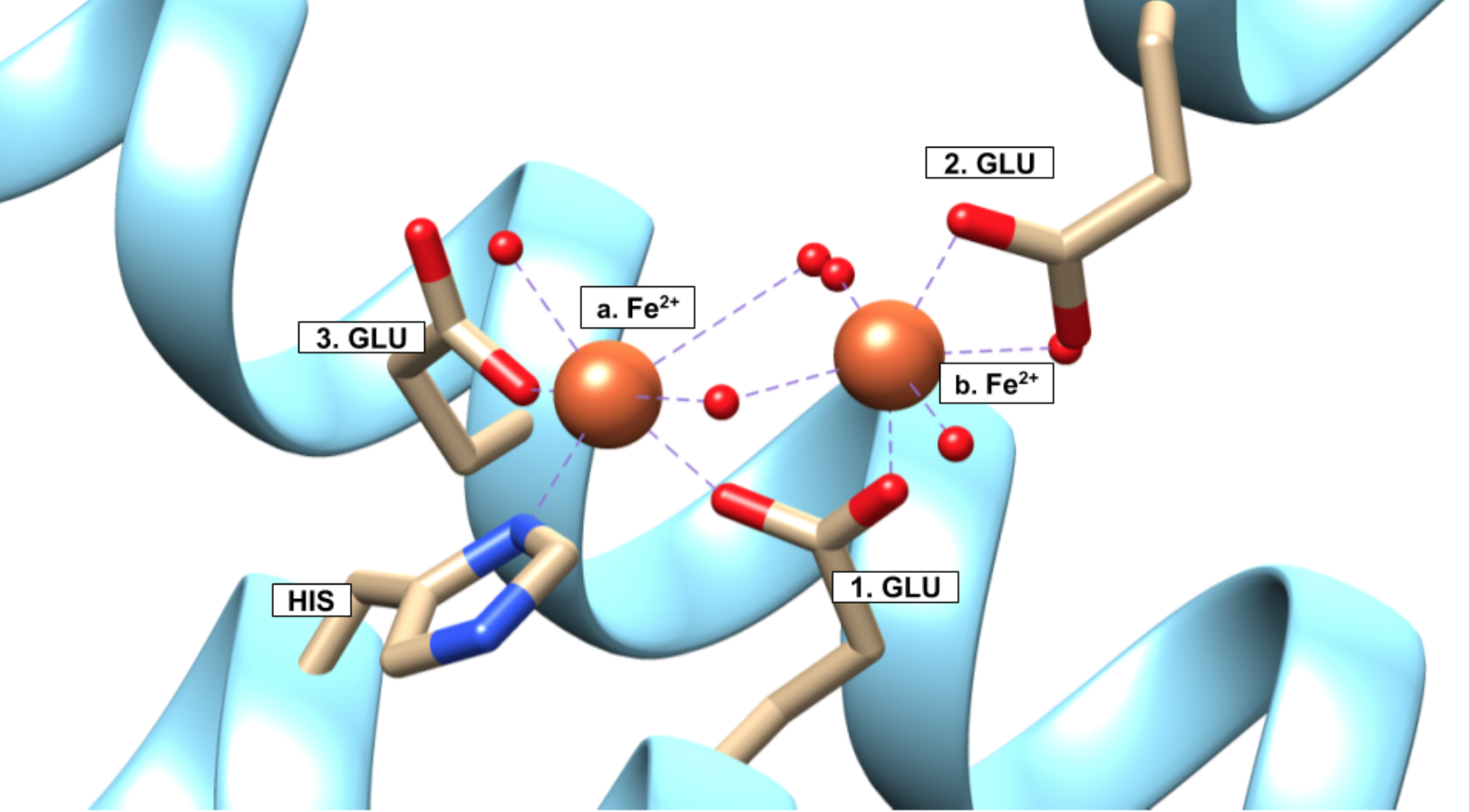
According to the crystal structure in Figure 6, the Fe2+ binds to a His residue and three different Glu residues all from the same protein. Ferritin can thus be considered a multidentate chelator for Fe2+. The ability for ferritin to perform as a chelator serves ferritin well. Because ferritin stores iron at such high volumes (up to 4500 iron ions), ferritin needs to be able to bind a lot of iron favorably. Based on the increase in entropy from binding, the chelate effect lets ferritin bind iron favorably. That ferritin can bind more than one metal ion to the same molecule creates an even greater increase in entropy and favorability.
Binding at the Ferroxidase Site
The human H-chain has two binding sites for iron(II) called site A and site B. Both sites are occupied by an iron ion, but site A has shown a higher affinity for iron(II).11 The mechanism for binding at the ferroxidase site is currently unknown. At the present moment, there are two common proposals: the stepwise mechanism and pairwise mechanism.1 The stepwise mechanism is shown in Figure 7. The mechanism proposes that the two binding sites are occupied one iron at a time.
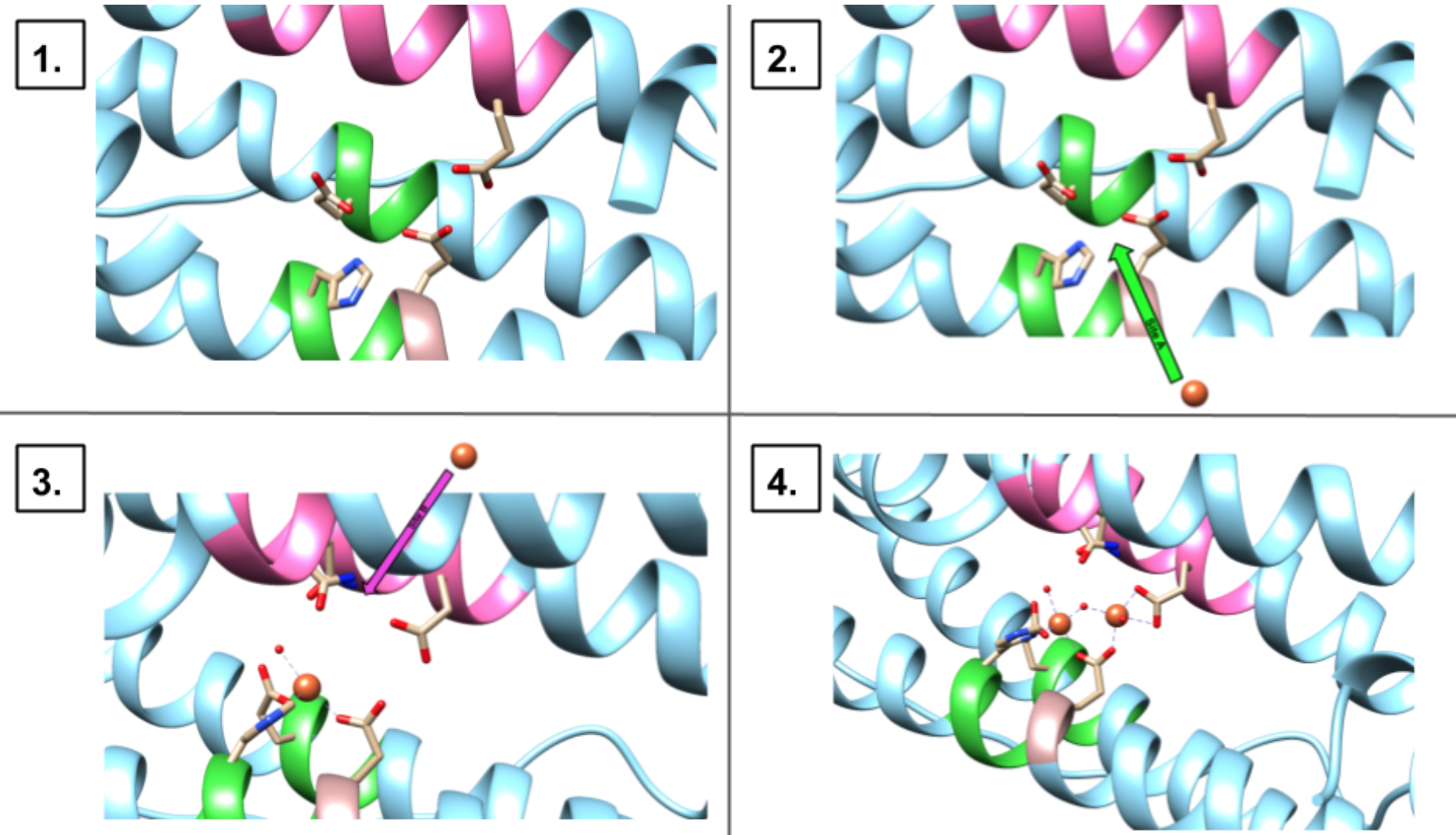
First, one Fe2+ binds to site A, then, a second Fe2+ will bind to site B. The pairwise mechanism shown in Figure 8 proposes that two Fe2+ ions enter site A and site B, in a concerted fashion.
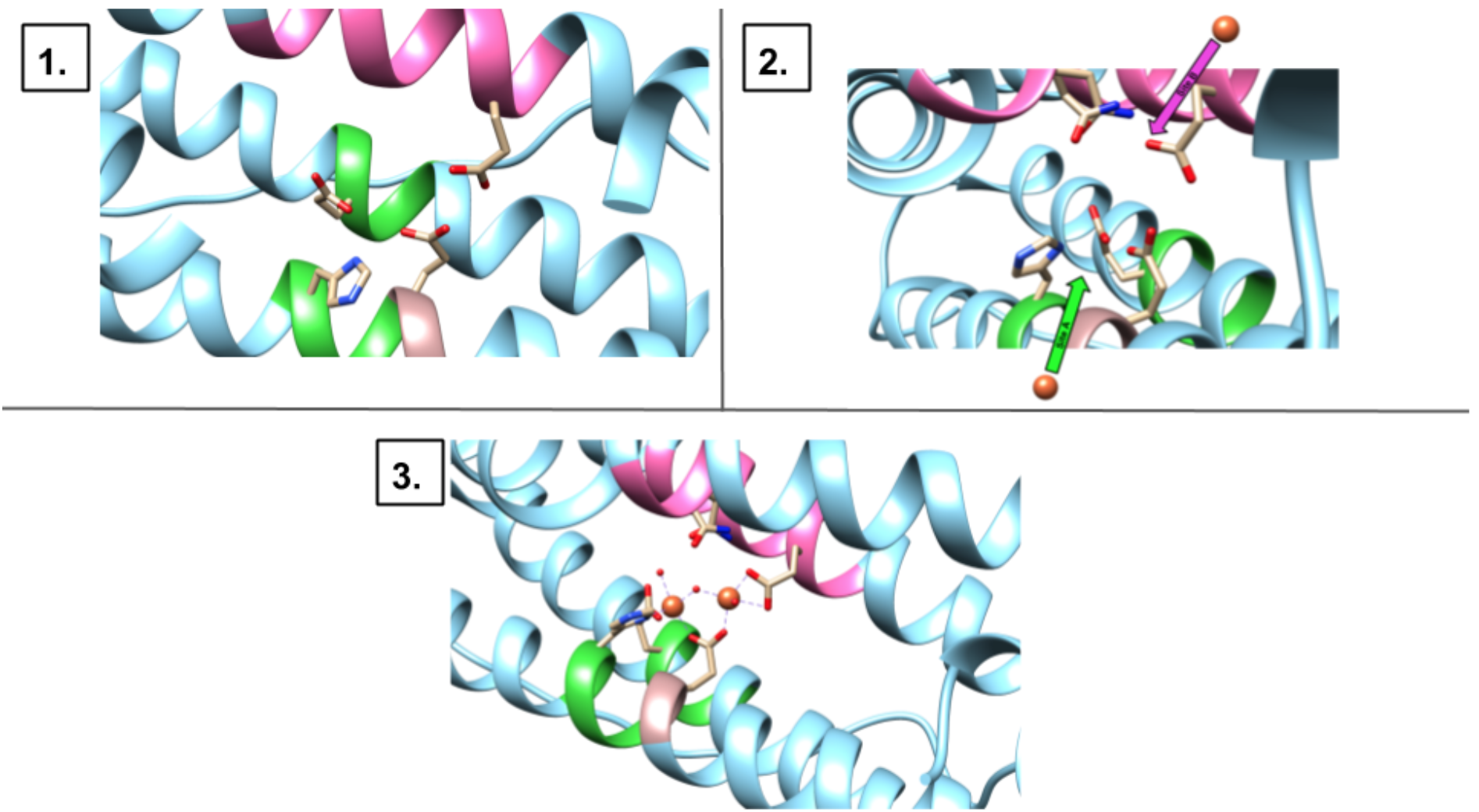 <
<
There has been evidence, however, that one site, site A, can be occupied by one Fe2+ while site B remains unoccupied. This result supports the stepwise mechanism (Figure 7) for binding at the ferroxidase site on the H-chain.1
After the iron ions bind to ferritin, a single molecule of some oxidizing agent binds to both ions. The binding of both ions by one molecule is referred to as bridging because the iron ions at sites A and B are bridged by the oxidizing agent.
Redox and Iron Mineralization
Iron(II) is oxidized at the ferroxidase site on the H-chain (sites A and B) by some oxidizing agent, most probably O2 and H2O2.1 Three reactions/models for Fe2+ oxidation and mineralization have been observed. There is a ferroxidation reaction or protein catalysis model (Reactions 1 and 1a-1c), a mineralization reaction or crystal growth model (Reaction 2), and a detoxification reaction of Fe2+ + H2O2 (Reaction 3).1 The ferroxidation reaction occurs at the ferroxidase site on the H-chain of ferritin. The ferroxidation reaction for the protein catalysis model of ferritin oxidation can be written as:
2 Fe2+ + O2 + 4 H2O → [Fe3+-O-O-Fe3+]2+ (1a.)
[Fe3+-O-O-Fe3+]2+ → [Fe2O-(OH)2]2+ (1b.)
[Fe2O(OH)2]2+ → 2FeOOHcore + H2O2 + 4 H+ (1c.)
Net: 2Fe2+ + O2 + 4 H2O → 2FeOOHcore + H2O2 + 4 H+ (1.).1
The net mineralization reaction for the crystal growth model of ferritin oxidation is written as:
4 Fe2+ + O2 + 6 H2O → 4 FeOOHcore + 8 H+ (2.).1
The most recently identified of the oxidation and mineralization reactions, the Fe2+ + H2O2 detoxification reaction is written as:
2 Fe2+ + H2O2 + 2 H2O → 2 FeOOHcore + 4 H+ (3.).1
The ferroxidation reaction involves the ferritin molecule most directly. Reaction 1 occurs on the H-chain at the ferroxidase site and has been the reaction focused on up to this point. The two schemes for this reaction that are most frequently proposed are a 2-electron oxidation scheme or a two successive 1-electron oxidation scheme.
The first intermediate produced in reaction 1a ([Fe3+-O-O-Fe3+]2+) is a μ-peroxo di-iron(III) complex (μ = number of metal ions bridged by a ligand, and μ2 is often shortened to just “μ”). Raman and Mössbauer spectroscopy, stopped-flow kinetics, and EXAFS have confirmed the existence of the complex. The complex is formed within the first 25-100 ms of the reaction and decays within 150 ms.1 The μ-peroxo di-iron(III) complex intermediate has been the focus of various studies for its blue coloring and appearance not only in human ferritin, but also other eukaryotic, archaeal, and bacterial ferritin.12 The blue intermediate from human H-chain ferritin has a UV-vis absorbance of 650 nm and an extinction coefficient measured at 850-1000 M-1 cm-1 per the dimer of [Fe3+-O-O-Fe3+]2+.1,12 The extinction coefficient (ε < 1000) suggests that this complex is absorbing d-d transitions.
The second intermediate is produced in reaction 1b ([Fe2O-(OH)2]2+) from the decay of the first intermediate is a μ-oxo(hydroxo)-bridged di-iron(III) intermediate.1 A distance of 3.00 Å has been observed between the two iron ions.11 Less data is available on this intermediate. While studies have provided strong evidence of the decay of the blue intermediate, the decay product is still in debate and the possibility of a variety of products has been noted.1
Oxidation of iron ions for storage in ferritin also proceeds through reactions 2 and 3. Studies suggest that the primary method and reaction of oxidizing iron for storage in ferritin depends on the flux of iron into ferritin at that time. For example, at a low flux of ≤ 50 Fe2+/ferritin, the iron is oxidized mainly by reaction 1 at the ferroxidase site.1 However, as more H2O2 is produced as a byproduct of reaction 1, the detoxification reaction (3) takes over as the primary reaction. Iron flux of about 100-500 Fe2+/ferritin will cause this switch.1 Once the iron flux reaches ~800 Fe2+/ferritin, reaction 2 becomes dominant.
Reaction 2 is based on the crystal growth model of ferritin oxidation. At high iron(II) flux into ferritin and after a minimum of 200 Fe2+/ferritin have already formed an iron/oxy mineral core in the center of ferritin, the crystal growth model appears to begin.1 In this model, iron(II) ions are entering at high rates. Rather than oxidizing every new iron(II) ion at the ferroxidase site or by the detoxification reaction, the iron(II) ions can be oxidized directly by the already-forming iron/oxy mineral core and can be deposited directly onto the mineral.1 As can be seen in reaction 2, stoichiometrically, iron(II) ions can be oxidized and directly deposited onto the mineral at 4 Fe2+/O2, while reaction 1 catalyzed by ferritin can only oxidize 2 Fe2+/O2.
It has not been determined if both Fe2+ ions are required to be bound at the ferroxidase site in order for oxidation to take place. However, unlike binding where there is evidence of a single Fe2+ ion being bound at site A, oxidation has never been observed without both Fe2+ ions, one at site A and one at site B and bridged by the oxidizing agent, present.1
As previously mentioned, nucleation starts on the L-chain subunit of ferritin. However, it is important to note that the majority of the actual iron/oxy mineral formed by the reactions above and held in the center of ferritin does not actually bind to the protein.8 Instead, the solid mineral core is stabilized by the protein surrounding it.8
Metal Center Geometries
The ligands for the ferroxidase site are His and Glu residues as well as a variety of oxygen-containing oxidants, such as O2 or H2O2 molecules.1,7 Ligand Field Theory (LFT) helps to predict the geometry of metal-ligand complexes. Complexes are classified as high spin or low spin based on the energy field splitting or Δo between the non-bonding t2g and the antibonding eg. High spin complexes will prefer to keep electrons unpaired in the t2g, but low spin complexes will prefer to pair electrons in the t2g. The ligands of LFT encourage strong fields (low spin, large Δo) or weak fields (high spin, small Δo) based on their categorization as a σ donor, π donor, or π acceptor.
The iron binding sites of ferritin, however, are not simple metal-ligand complexes. As discussed above, when binding to the ferroxidase, two ions of iron(II) bind to the site and are bridged by the oxidizing agent, most commonly O2. The bridging ligand makes this one binding site and one complex, not two separate single metal or mononuclear complexes. The ferroxidase site is considered to have a di-iron center and to be a dinuclear binding site or dinuclear complex.1 Because there are two metals in the metal center, molecular orbital interactions in dinuclear complexes are different than those in the one metal center. This difference is especially noticed in bridged metal complexes, as is present in ferritin, because the orbitals of the two metal ions can overlap.13 Due to this overlap, electron filling and pairing does not always occur as one would expect with a single metal center.
These dinuclear bridged complexes with iron(II) and iron(III) are not unique to ferritin, but have been seen in methane monooxygenase (MMO), ribonucleotide reductase (RNR), and fatty acid desaturase.8,12 Comparisons with these other proteins with ferritin have been attempted, but iron’s special role as a substrate in ferritin rather than an enzyme cofactor in the others sets it apart and makes ferritin more difficult to discern.12 Furthermore, the comparisons with these other proteins is not completely correct based on the lability of the di-iron complexes in ferritin. The di-iron(II) binding site complex and the di-iron(III) intermediates of oxidation are labile as they react, degrade, and move toward the center of ferritin. In contrast, the di-iron complexes of MMO are stable cofactors.
For ease, ferritin’s ferroxidase binding site will be analyzed according to LFT without consideration of the overlapping of molecular orbitals. Note though that the following analysis does not entirely capture the complexity of the metal-ligand-metal interactions.
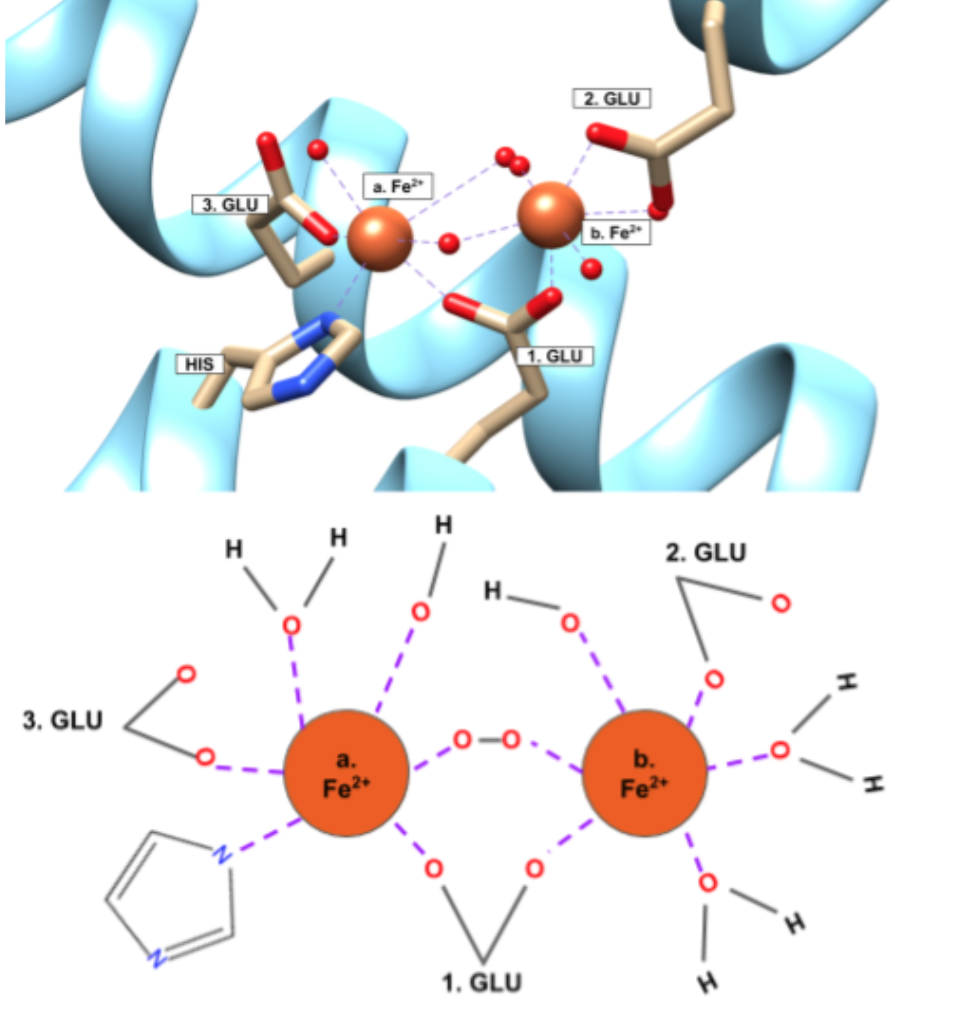
According to LFT, His residue is a π acceptor and a weak field ligand. This ligand increases Δo and encourages low spin complexes. All oxygen ligands, including water, OH-, O2-, and Glu residues are π donors and weak field ligands, which decrease Δo and encourage high spin complexes. A close-up of the binding sites at the ferroxidase site, such as in Figure 7, shows that the majority of the ligands for both a. Fe2+ and b. Fe2+ are water or the like and Glu residues, the high spin complex-favoring ligands. Fe2+ is a 3d6 transition metal with a charge of +2 and thus can be high or low spin. However, when bound to H2O, Fe2+ is high spin with an electron filling diagram shown in Figure 9.
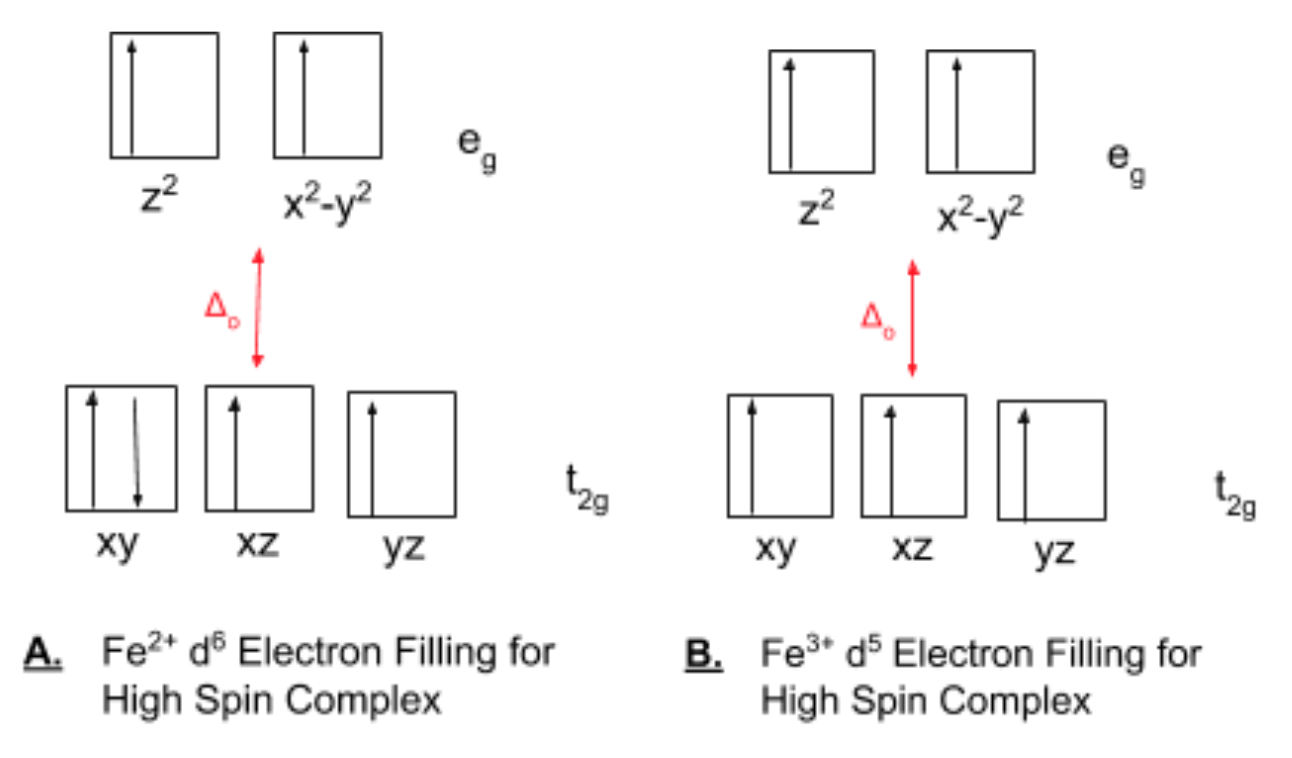
The complex is thus, overall, high spin and has been described in the literature as initially binding in a high-spin state with octahedral geometry shown in Figure 10.11,14
The ligands for the iron oxide mineral are primarily H2O, OH-, or O2-. These use the oxygen as the donor atom. These are all high spin ligands that contribute to the weak field. These ligands will favor a smaller Δo and keeping electrons unpaired. Fe3+ is a 3d5 transition metal with a +3 charge and is more likely to be low spin based on recorded, however, known values of Δo for Fe3+ and literature on the Fe3+ intermediates categorize these Fe3+-O-Fe3+ groups as high spin (see electron filling diagram in Figure 9 for a single metal center high spin diagram). Δowill thus be smaller and weak field ligands such as the oxygen ligands will be favored.
To highlight the complexity of the dinuclear metal-ligand complex, studies on the magnetic susceptibility (the ratio of magnetic moment per unit volume to the applied magnetizing field intensity) of the Fe3+-O-Fe3+ groups suggest that the high spin iron complexes have about one unpaired electron per group.14 This calculation is clearly in conflict with the molecular orbital diagram in Figure 9, yet the complex is still known to be on average octahedral and high spin.14 The calculation magnetic moment itself has been a controversial pursuit among scientists.9
Iron Release
When cells require iron, for enzyme synthesis, after blood loss, or during embryonic development, the iron stored in ferritin must be released rapidly, on demand, and under control.8 Although, as with many aspects of ferritin function, the mechanism of iron release to the cell from ferritin is unknown. What is known inherently about the process is that over its course it must essentially reverse mineralization: the iron(III)/oxy mineral must be dissolved from its solid state to aqueous ions and the iron(III) ions must be reduced to iron(II).
Proposed models for ferritin iron release include (1) an equilibrium between the iron stored in ferritin and the iron in the cytoplasm of the cell, (2) ferritin protein degradation, (3) spontaneous, direct dissolution of iron(III) from the mineral core from scavenging by iron(III) binding proteins, and (4) the reduction of the iron(III) mineral which is then complexed iron(II) by a chelating agent and transported out of ferritin.4,12 The last model (4) lacks confirmation, yet is considered by many to be the physiological mechanism. This model is the most efficient method and the presence of reducing agents under physiological conditions.4
Except the second model (2), the physiological relevance of all above models is still to be seen.12 The ferritin degradation model (2) is the only mechanism to have been observed under physiological conditions. However, since this model would require iron release to be dependent of the turnover and synthesis of ferritin, another model is suspected to be necessary.12
However iron is released from ferritin, studies indicate that the physical exit of the iron from ferritin takes place via the 3-fold channels. One proposal for exit based on observations in bacterial ferritin is the localized folding/unfolding of ferritin.4 The ferritin channels can be unfolded without affecting the overall function or structure of the protein. Unfolding the channels and thus, widening the opening, would allow for quick release of demineralized iron. The channels become highly disordered when this occurs to the effect that they do not appear in crystal structures.8
Much remains to be determined about human ferritin. Other functions of ferritin in the body, and the mechanisms for how Fe2+ enters and binds to ferritin, how Fe2+ is oxidized to Fe3+ for storage, and how iron is released remain undetermined, among others, are thought to be non-universal—even within the same organism.3,6
While much of the chemistry behind ferritin is, as of now, unexplained, ferritin is still an important protein for all life. Wherever iron is noted as an essential nutrient, ferritin must also be present for management and storage. The elucidation of ferritin can lead to advancements in iron metabolism and neurological disorders and new uses of ferritin chemistry in nanochemistry and catalytic industrial applications.3,4
Sources
[1] Bou-Abdallah, F. The Iron Redox and Hydrolysis Chemistry of the Ferritins. Biochimica et Biophysica Acta (BBA) - General Subjects 2010, 1800 (8), 719–731.
[2] Crabb, E.; Moore, E. Metals and life; Royal Society of chemistry: Cambridge, 2010.
[3] Theil, E. C.; Tosha, T.; Behera, R. K. Solving Biology’s Iron Chemistry Problem with Ferritin Protein Nanocages. Accounts of Chemical Research 2016, 49 (5), 784–791.
[4] Carmona, F.; Palacios, Ò.; Gálvez, N.; Cuesta, R.; Atrian, S.; Capdevila, M.; Domínguez-Vera, J. M. Ferritin Iron Uptake and Release in the Presence of Metals and Metalloproteins: Chemical Implications in the Brain. Coordination Chemistry Reviews 2013, 257 (19–20), 2752–2764.
[5] Knovich, M. A.; Storey, J. A.; Coffman, L. G.; Torti, S. V. Ferritin for the Clinician. Blood Rev 2009, 23 (3), 95–104.
[6] Bradley, J. M.; Le Brun, N. E.; Moore, G. R. Ferritins: Furnishing Proteins with Iron. JBIC Journal of Biological Inorganic Chemistry 2016, 21 (1), 13–28.
[7] Watt, G. D.; Jacobs, D.; Frankel, R. B. Redox Reactivity of Bacterial and Mammalian Ferritin: Is Reductant Entry into the Ferritin Interior a Necessary Step for Iron Release? Proceedings of the National Academy of Sciences 1988, 85 (20), 7457–7461.
[8] Bertini, I. Biological Inorganic Chemistry: Structure and Reactivity; University Science Books, 2007.
[9] Chasteen, N. D.; Harrison, P. M. Mineralization in Ferritin: An Efficient Means of Iron Storage. Journal of Structural Biology 1999, 126 (3), 182–194.
[10] Joshi, J. G.; Sczekan, S. R.; Fleming, J. T. Ferritin—A General Metal Detoxicant. Biol Trace Elem Res 1989, 21 (1), 105.
[11] Bertini, I.; Lalli, D.; Mangani, S.; Pozzi, C.; Rosa, C.; Theil, E. C.; Turano, P. Structural Insights into the Ferroxidase Site of Ferritins from Higher Eukaryotes. Journal of the American Chemical Society 2012, 134(14), 6169–6176.
[12] Honarmand Ebrahimi, K.; Hagedoorn, P.-L.; Hagen, W. R. Unity in the Biochemistry of the Iron-Storage Proteins Ferritin and Bacterioferritin. Chemical Reviews 2015, 115 (1), 295–326.
[13] H. Rodriguez, J.; Mccusker, J. Density Functional Theory of Spin-Coupled Models for Diiron-Oxo Proteins: Effects of Oxo and Hydroxo Bridging on Geometry, Electronic Structure, and Magnetism. Journal of Chemical Physics 2002, 116.
[14] Bertini, I., Bioinorganic Chemistry; Ed.; University Science Books: Mill Valley, Calif, 1994.
Contributed By
This work was originally written by Anna Shadid, Spring 2018: Anna is currently (as of 2018) a junior chemistry major at Saint Mary's College in Notre Dame, IN.
This work was originally edited by Dr. Kathryn Haas, Associate Professor, and Madison Sendzik, Teaching and Research Assistant, of Saint Mary's College.