
14.3: Substituent Effects
- Page ID
- 225862
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Activation and Deactivation
Because benzene acts as a nucleophile in electrophilic aromatic substitution, substituents that make the benzene more electron-rich can accelerate the reaction. Substituents that make the benzene moor electron-poor can retard the reaction. In the mid-twentieth century, physical organic chemists including Christopher Ingold conducted a number of kinetic studies on electrophilic aromatic substitution reactions. In table 1, you can see that some substituents confer a rate of reaction that is much higher than that of benzene (R = H). Phenol, C6H5OH, undergoes nitration a thousand times faster than benzene does. Nitrobenzene, C6H5NO2, undergoes the reaction millions of times more slowly.
| R in C6H5R | Relative rate |
| OH | 1,000 |
| CH3 | 25 |
| H | 1 |
| CH2Cl | 0.71 |
| I | 0.18 |
| F | 0.15 |
| Cl | 0.033 |
| Br | 0.030 |
| CO2Et | 0.0037 |
| NO2 | 6 x 10-8 |
| NMe3+ | 1.2 x 10-8 |
These observations are consistent with the role of the aromatic as a nucleophile in this reaction. Substituents that draw electron density away from the aromatic ring slow the reaction down. These groups are called deactivating groups in this reaction. Substituents that readily donate electron desnity to the ring, or that effectively stabilize the cationic intermediate, promote the reaction. These groups are called activating groups in this reaction.
The roles of these groups are related to their electronic interactions with the electrons in the ring. Some groups might be π-donors, providing additional electron density to the benzene ring via resonance.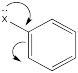
Other groups may be π-acceptors, drawing electron density away from the ring via resonance.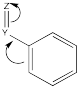
Still others may be σ-acceptors, drawing electron density away from the ring via a simple inductive effect which arises from the electronegativity of a substituent.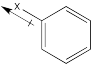
In some cases, there may be multiple effects, and the overall influence of the substituents is determined by the balance of the effects. One effect may be stronger in one case than the other, so it wins out in one case and loses in another.
Directing Effects
In addition to exerting an effect on the speed of reaction, substituents on the benzene ring also influence the regiochemistry of the reaction. That is, they control where the new substituent appears in the product.
Remember, there are three different positions on the benzene ring where a new substituent can attach, relative to the original substituent. Substitution could actually occur on five positions around the ring, but two pairs are related by symmetry. Isomerism in disubstituted benzenes can be described by numbering the substituents (1,2- etc) or by the relationships ortho-, meta– and para-. There are two positions ortho– to the initial substituent and two positions meta– to it.
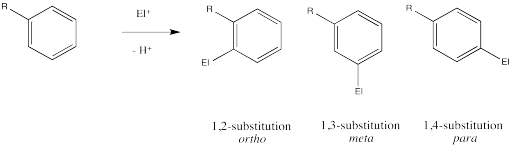
Ingold and colleagues investigated the question of regiochemistry in nitration. They reported the following observations:
| R in C6H5R | % o– product | % m– product | % p– product |
| CH3 | 56 | 3 | 41 |
| Cl | 30 | 0 | 70 |
| Br | 38 | 0 | 62 |
| OH | 10 | 0 | 90 |
| CHO | 19 | 72 | 9 |
| CO2Et | 28 | 68 | 3 |
| CN | 17 | 81 | 2 |
| NO2 | 6 | 94 | 0 |
In looking at the table, you might see that there are two groups of substituents. One group reacts to make mixtures of ortho– and para– products. There may be different ratios of ortho– to para– and there may be small amounts of meta-, but don’t get bogged down in the details. Focus on the bigger picture. Some groups are “ortho/para directors”.
The other group reacts to make mostly meta-substituted products. Here may be small amounts of ortho– and para– products, but these groups are best described as “meta-directors”. These regiochemical effects are very closely related to the activating and directing effects we have already seen. If we want to understand these data, we need to think about things like π-donation, π-acceptance, inductive effects and cation stability.
Explaining directing effects in Friedel-Crafts reactions
The reactivity of aromatic pi bonds in SEAr reactions is very sensitive to the presence of electron-donating groups (EDG) and electron-withdrawing groups (EWG) on the aromatic ring. This is due to the carbocation nature of the intermediate, which is stabilized by electron-donating groups and destabilized by electron-withdrawing groups.
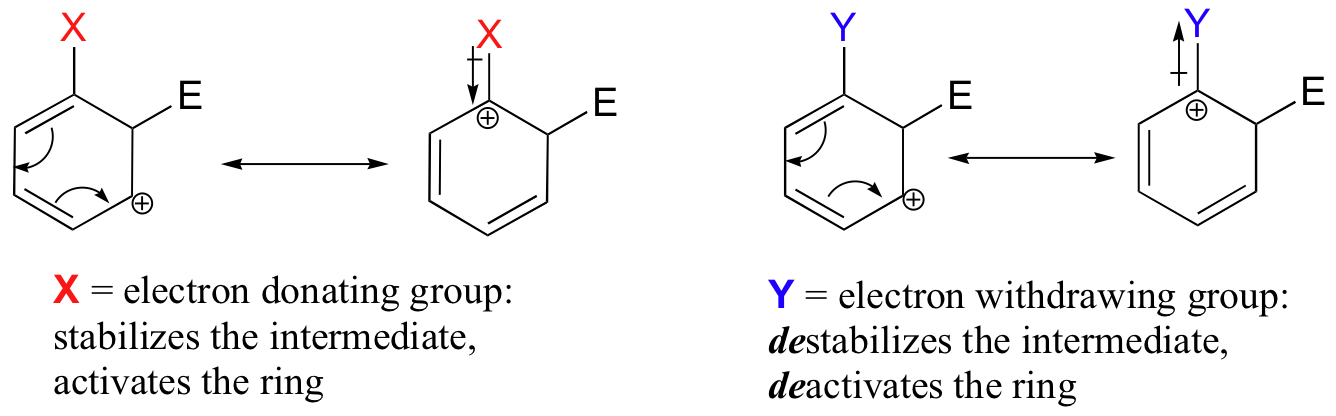
Alkyl groups are weakly ring-activating groups, as their electron-donating ability stems only from weak inductive effects. Substituents with heteroatoms connected to the aromatic ring are significantly more ring-activating than alkyl groups, because resonance electron-donating effects are possible. Amines, for example, are very powerful ring-activating substituents, due to the ability of the lone pair on the nitrogen to stabilize the carbocation intermediate through resonance:

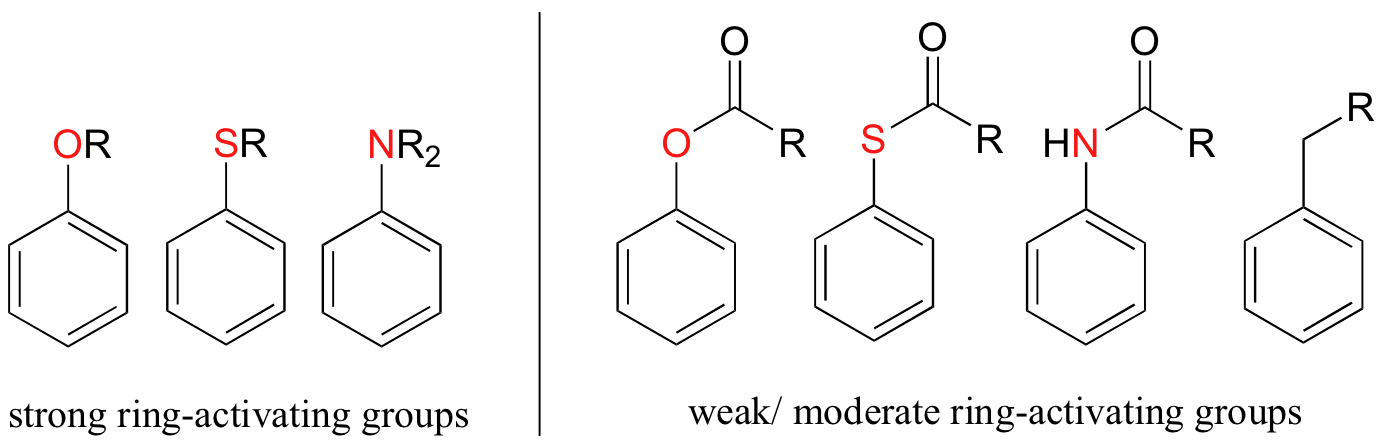
Other ring-activating groups are shown below (in these figures, the R group can be a hydrogen). All of these groups are able, in varying degrees, to stabilize the carbocation intermediate in an electrophilic aromatic substitution reaction. Notice that plain old alkyl groups are also (weakly) ring-activating.
Substituent groups that are ring-activating due to resonance effects also tend to exert a strong regiochemical influence on further substitution reactions. Specifically, substitution tends to occur in the ortho and para positions relative to the existing group. This is known as the ortho-para directing effect. The effect can be explained by drawing resonance contributors for the carbocation intermediate of the SEAr reaction: the positive charge is in position to be delocalized by resonance only in reactions leading to ortho or para substitution.

The carbocation which leads to the meta-substituted product, however, cannot be stabilized by resonance with the ring-activating group:

As an example, the Friedel-Crafts alkylation of methoxy benzene would be expected to produce a mixture of the ortho and para substituted products, but no meta-substituted product.
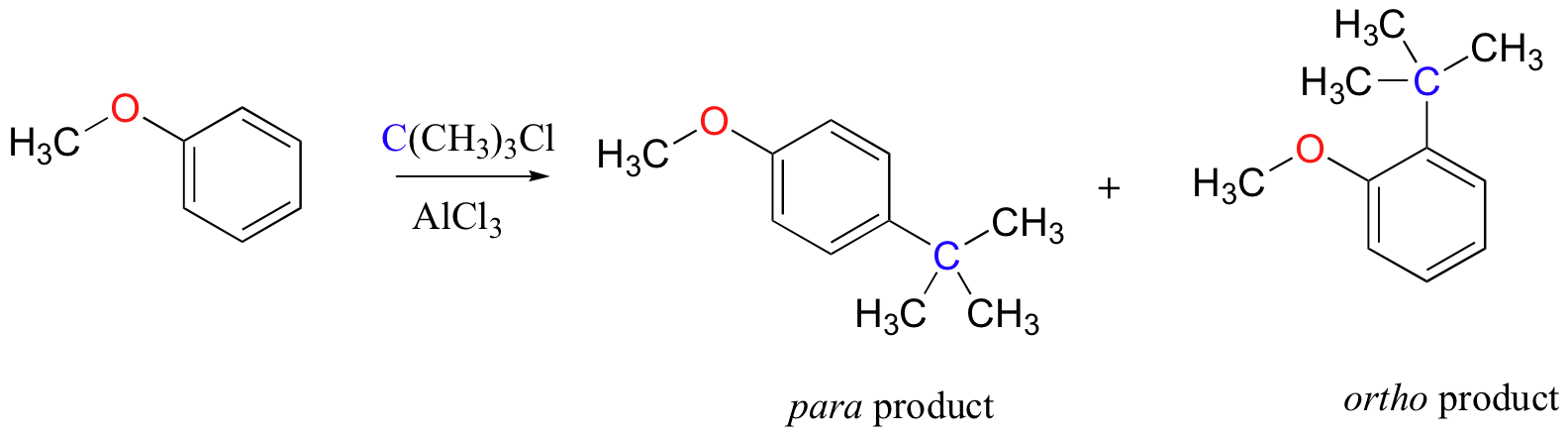
In addition, the para product would be expected to be preferred over the ortho product, due to steric considerations.
Electron-withdrawing substituents on an aromatic ring are ring-deactivating, making it harder for further substitution reactions to occur. These are mostly carbonyl-containing groups, as well as alkyl halides.
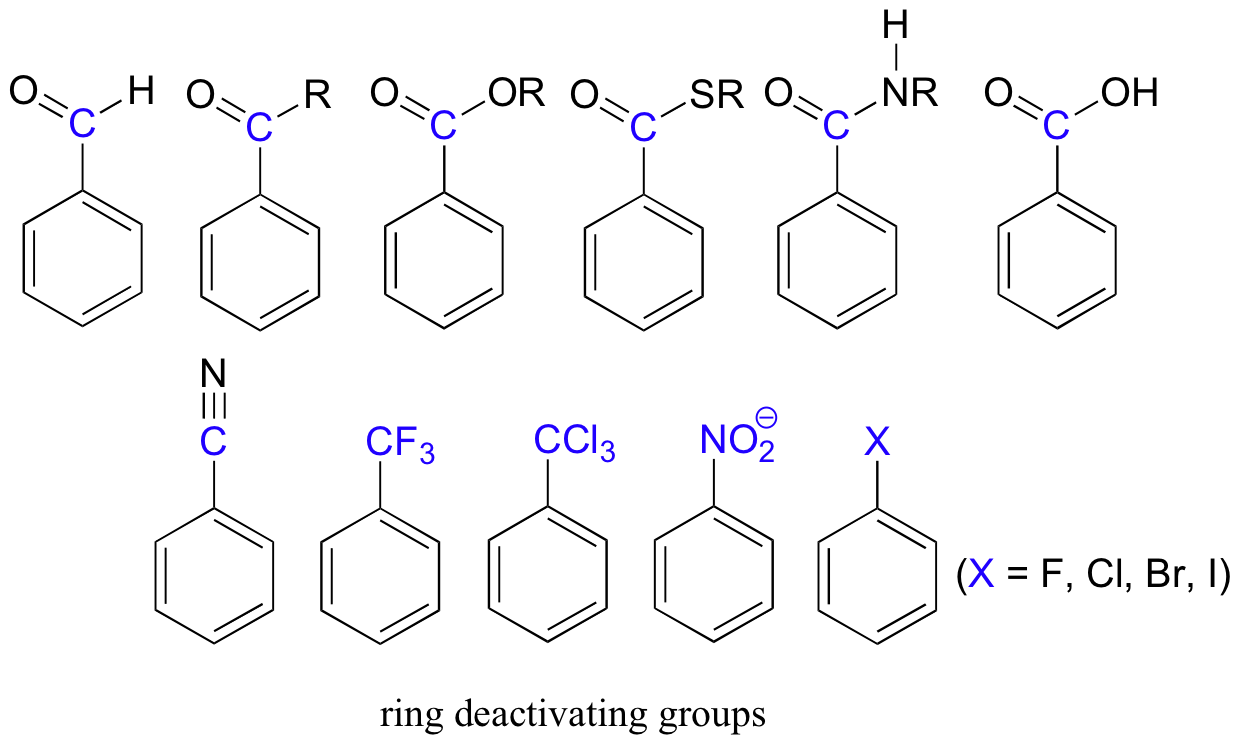
When substitution does occur on an aromatic ring with deactivating group already attached, it tends to occur specifically at the meta position – deactivating groups are generally meta-directing. The exception to this rule is the halogens, which are ring-deactivating but ortho-para directing (see next section).
Halogens: A Special Case
Halogens are very electronegative. This means that inductively they are electron-withdrawing. However, because of their ability to donate a lone pair of electrons in resonance forms, they are ortho/para directing. Resonance effects win out in directing ortho-para, but the inductive effect is stronger in determining the reactivity: Because (on balance) they are electron withdrawing, halogens are very weak activators.
Directing effects of some common substituents
| Orthe- and Para- Directing | Meta Directing | ||||
|---|---|---|---|---|---|
| Strong Activating | Moderately Activating | Weakly Activating | Weakly Deactivating | Moderately Deactivating | Strongly Deactivating |
| -NH2 -NHR -OH -OCH3 | -NHCOR -OCOR | -CH3 -phenyl | -F -Cl -Br -I | -COH -COCH3 -COOCH3 -SO3H | -NO2 -CF3 -CCl3 |
References
- Bohm, S., and O. Exner. “Interaction of two functional groups through the benzene ring: Theory and Experiment.” Journal of Computational Chemistry (2008) (p. 1) http://www.lib.berkeley.edu
- Brown, William H., Foote, Christopher S., Iverson, Brent L. Organic Chemisty. 4th ed. Belmont, CA: Thomson Learning Inc./ Brooks/Cole, 2005. (pp. 868-872)
- Schore, Neil E., Vollhardt, Peter C. Organic Chemistry, Structure and Function. 5th ed. New York: W.H. Freeman & Company, 2007. (pp. 724-728)
- Laali, Kenneth K., and Volkar J. Gettwert. “Electrophilic Nitration of Aromatics in Ionic Liquid Solvents.” The Journal of Organic Chemistry 66 (Dec. 2000): 35-40. American Chemical Society.
- Malhotra, Ripudaman, Subhash C. Narang, and George A. Olah. Nitration: Methods and Mechanisms. New York: VCH Publishers, Inc., 1989.
- Sauls, Thomas W., Walter H. Rueggeberg, and Samuel L. Norwood. “On the Mechanism of Sulfonation of the Aromatic Nucleus and Sulfone Formation.” The Journal of Organic Chemistry 66 (1955): 455-465. American Chemical Society.
- Vollhardt, Peter. Organic Chemistry : Structure and Function. 5th ed. Boston: W. H. Freeman & Company, 2007.
Exercises
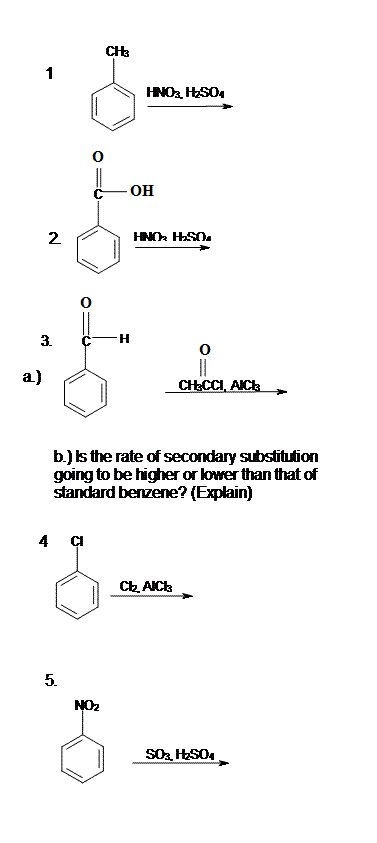
Solutions
[reveal-answer q=”862551″]Show Answer[/reveal-answer]
[hidden-answer a=”862551″]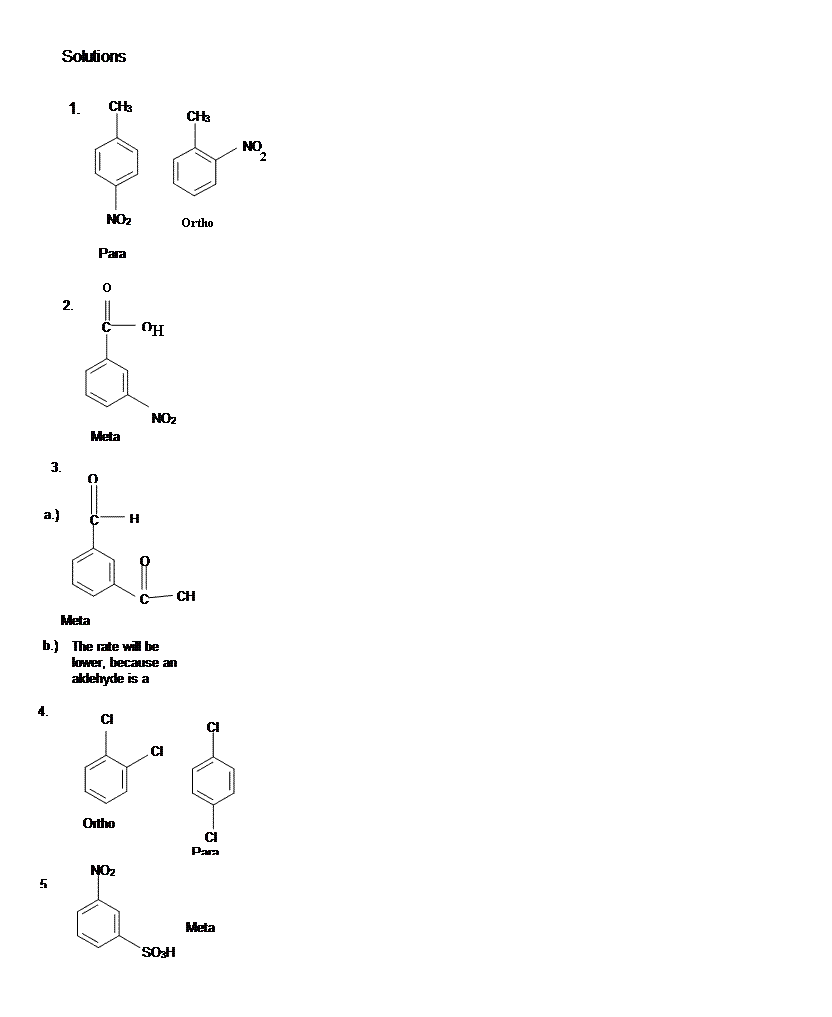
[/hidden-answer]
Outside Links
Aromatic Sulfonation
- Wikipedia: http://en.wikipedia.org/wiki/Aromatic_sulfonation
- Video: http://www.youtube.com/watch?v=s1qJ1…eature=related
- Interactive 3D Reaction: http://www.chemtube3d.com/Electrophi…20benzene.html
Aromatic Nitration
- Wikipedia: http://en.wikipedia.org/wiki/Nitration
- Video: http://www.youtube.com/watch?v=i7ucl…eature=related
- Interactive 3D Reaction: http://www.chemtube3d.com/Electrophi…20benzene.html
Contributors
- Chris P Schaller, Ph.D., (College of Saint Benedict / Saint John’s University)
- Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)
- Lauren Rice and Samantha Spragg (UCD)
- 27.6: Electrophilic Aromatic Substitution. Authored by: Chris P. Schaller. Provided by: Chemistry LibreTexts. Located at: https://chem.libretexts.org/Textbook_Maps/General_Chemistry/Map%3A_General_Chemistry_(Petrucci_et_al.)/27%3A_Reactions_of_Organic_Compounds/27.06%3A_Electrophilic_Aromatic_Substitution. Project: General Chemistry. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 15.6: Synthetic parallel - electrophilic aromatic substitution in the lab. Authored by: Tim Soderberg. Provided by: Chemistry LibreTexts. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/15%3A_Electrophilic_reactions/15.06%3A_Synthetic_parallel_-_electrophilic_aromatic_substitution_in_the_lab. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- Inductive Effects of Alkyl Groups. Authored by: Lauren Rice and Samantha Spragg . Provided by: Chemistry LibreTexts. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Arenes/Properties_of_Arenes/Inductive_Effects_of_Alkyl_Groups. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 18.4: Nitration and Sulfonation. Authored by: Anonymous. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Map%3A_Organic_Chemistry_(Smith)/Chapter_18%3A_Electrophilic_Aromatic_Substitution/18.4%3A_Nitration_and_Sulfonation. Project: Chemistry LibreText. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike