
27.6: Electrophilic Aromatic Substitution
- Page ID
- 24393
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Aromatics, or arenes, are derivatives of benzene or other compounds with aromatic ring systems. That is, they are cyclic, planar, fully conjugated and have an odd number of π-electron pairs. Like alkenes, aromatics have π-electrons that are loosely held and are easily attracted to electrophiles. However, aromatics don't undergo the typical reactions of alkenes.
For example, bromine will not add across the double bond of benzene.

Instead, a bromine atom replaces one of the hydrogen atoms on the benzene. This reaction is greatly accelerated in the presence of Lewis acids, such as ferric chloride.

A similar reaction happens with chlorine. If treated with chlorine gas and a metal catalyst, a chlorine atom from chlorine gas can replace a hydrogen atom on benzene. However, the same thing doesn't work as smoothly with the other halogens, iodine and fluorine.
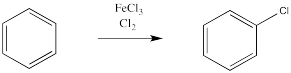
The reactions of chlorine and bromine with benzene and other aromatics can be catalysed by a variety of Lewis acidic metal catalysts. So can the reactions of alkyl halides and acyl halides, which we don't normally think of as electrophiles for alkene addition.

There are some limitations on what kind of groups can be added in this way. The carbon attached to the halide should be tetrahedral. Typically, it is much easier to add secondary or tertiary alkyls than primary ones. That is, the carbon attached to the halogen had best be attached to two or three other carbons as well. Methyls are very, very difficult to add in this way.
There is an exception - the carbon attached to the halogen need not be tetrahedral, provided it is a carbonyl carbon. That reaction is called an acylation.
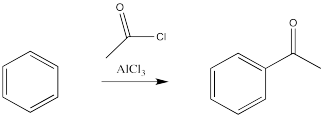
In these cases, it is the alkyl or acyl, rather than the halogen, that replaces a hydrogen atom on the benzene. Remember, benzene is most likely acting as a nucleophile in this reaction, even though it is following a different pathway than an alkene would. It is reacting with the most electrophilic part of the alkyl halide or acyl halide.
Aromatics have a limited repertoire of electrophiles with which they commonly undergo reaction. In addition to these Lewis acid-catalysed reactions, there are also reactions strong acidic media, such as a mixture of nitric and sulfuric acid.
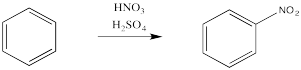
Another acidic medium, referred to as "fuming sulfuric acid," is really a mixture of sulfuric acid and sulfur trioxide.
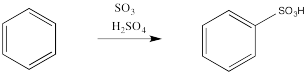
Just as with the acid-catalyzed reactions, the nitro group and the sulfonate group just replace a hydrogen atom on the benzene ring. The overall reaction involves bond formation between a benzene carbon and the electrophile, and bond cleavage between the same carbon and a proton.
Activation and Deactivation and Directing Effects
Because the benzene acts as a nucleophile in electrophilic aromatic substitution, substituents that make the benzene more electron-rich can accelerate the reaction. Substituents that make the benzene moor electron-poor can retard the reaction. In the mid-twentieth century, physical organic chemists including Christopher Ingold conducted a number of kinetic studies on electrophilic aromatic substitution reactions. In table 1, you can see that some substituents confer a rate of reaction that is much higher than that of benzene (R = H). Phenol, C6H5OH, undergoes nitration a thousand times faster than benzene does. Nitrobenzene, C6H5NO2, undergoes the reaction millions of times more slowly.
| R in C6H5R | Relative rate |
| OH | 1,000 |
| CH3 | 25 |
| H | 1 |
| CH2Cl | 0.71 |
| I | 0.18 |
| F | 0.15 |
| Cl | 0.033 |
| Br | 0.030 |
| CO2Et | 0.0037 |
| NO2 | 6 x 10-8 |
| NMe3+ | 1.2 x 10-8 |
These observations are consistent with the role of the aromatic as a nucleophile in this reaction. Substituents that draw electron density away from the aromatic ring slow the reaction down. These groups are called deactivating groups in this reaction. Substituents that readily donate electron desnity to the ring, or that effectively stabilize the cationic intermediate, promote the reaction. These groups are called activating groups in this reaction.
The roles of these groups are related to their electronic interactions with the electrons in the ring. Some groups might be π-donors, providing additional electron density to the benzene ring via conjugation.
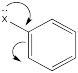
Other groups may be π-acceptors, drawing electron density away from the ring via conjugation.
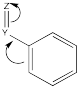
Still others may be σ-acceptors, drawing electron density away from the ring via a simple inductive effect which arises from the electronegativity of a substituent.
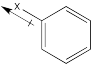
In some cases, there may be multiple effects, and the overall influence of the substituents is determined by the balance of the effects. One effect may be stronger in one case than the other, so it wins out in one case and loses in another.
In addition to exerting an effect on the speed of reaction, substituents on the benzene ring also influence the regiochemistry of the reaction. That is, they control where the new substituent appears in the product.
Remember, there are three different position on the bezene ring where a new substituent can attach, relative to the original substituent. Substitution could actually occur on five positions around the ring, but two pairs are related by symmetry. Isomerism in disubstituted benzenes can be described by numbering the substituents (1,2- etc) or by the relationships ortho-, meta- and para-. There are two positions ortho- to the initial substituent and two positions meta- to it.
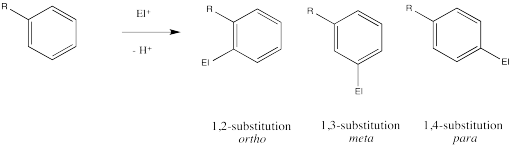
Ingold and colleagues investigated the question of regiochemistry in nitration. They reported the following observations:
| R in C6H5R | % o- product | % m- product | % p- product |
| CH3 | 56 | 3 | 41 |
| Cl | 30 | 0 | 70 |
| Br | 38 | 0 | 62 |
| OH | 10 | 0 | 90 |
| CHO | 19 | 72 | 9 |
| CO2Et | 28 | 68 | 3 |
| CN | 17 | 81 | 2 |
| NO2 | 6 | 94 | 0 |
In looking at the table, you might see that there are two groups of substituents. One group reacts to make mixtures of ortho- and para- products. There may be different ratios of ortho- to para- and there may be small amounts of meta-, but don't get bogged down in the details. Focus on the bigger picture. Some groups are "ortho-/para-directors".
The other group reacts to makemostly meta-substituted products. Here may be small amounts ofortho- and para- products, but don't worry about that. Focus on the bigger picture. Some groups are "meta-directors". These regiochemical effects are very closely related to the activating and directing effects we have already seen. If we want to understand this data, we need to think about things like π-donation, π-acceptance, inductive effects and cation stability.