
9.3: Enthalpy
- Page ID
- 111895

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- State the first law of thermodynamics
- Define enthalpy and explain its classification as a state function
- Write and balance thermochemical equations
- Calculate enthalpy changes for various chemical reactions
- Explain Hess’s law and use it to compute reaction enthalpies
Thermochemistry is a branch of chemical thermodynamics, the science that deals with the relationships between heat, work, and other forms of energy in the context of chemical and physical processes. As we concentrate on thermochemistry in this chapter, we need to consider some widely used concepts of thermodynamics.
Substances act as reservoirs of energy, meaning that energy can be added to them or removed from them. Energy is stored in a substance when the kinetic energy of its atoms or molecules is raised. The greater kinetic energy may be in the form of increased translations (travel or straight-line motions), vibrations, or rotations of the atoms or molecules. When thermal energy is lost, the intensities of these motions decrease and the kinetic energy falls. The total of all possible kinds of energy present in a substance is called the internal energy (U), sometimes symbolized as E.
As a system undergoes a change, its internal energy can change, and energy can be transferred from the system to the surroundings, or from the surroundings to the system. Energy is transferred into a system when it absorbs heat (q) from the surroundings or when the surroundings do work (w) on the system. For example, energy is transferred into room-temperature metal wire if it is immersed in hot water (the wire absorbs heat from the water), or if you rapidly bend the wire back and forth (the wire becomes warmer because of the work done on it). Both processes increase the internal energy of the wire, which is reflected in an increase in the wire’s temperature. Conversely, energy is transferred out of a system when heat is lost from the system, or when the system does work on the surroundings.
The relationship between internal energy, heat, and work can be represented by the equation:
\[ΔU=q+w \label{5.4.1} \]
as shown in Figure \(\PageIndex{1}\). This is one version of the first law of thermodynamics, and it shows that the internal energy of a system changes through heat flow into or out of the system (positive q is heat flow in; negative q is heat flow out) or work done on or by the system. The work, w, is positive if it is done on the system and negative if it is done by the system.
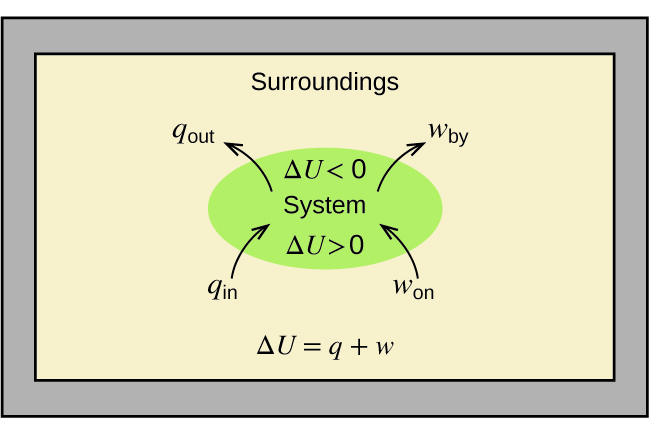
A type of work called expansion work (or pressure-volume work) occurs when a system pushes back the surroundings against a restraining pressure, or when the surroundings compress the system. An example of this occurs during the operation of an internal combustion engine. The reaction of gasoline and oxygen is exothermic. Some of this energy is given off as heat, and some does work pushing the piston in the cylinder. The substances involved in the reaction are the system, and the engine and the rest of the universe are the surroundings. The system loses energy by both heating and doing work on the surroundings, and its internal energy decreases. (The engine is able to keep the car moving because this process is repeated many times per second while the engine is running.) We will consider how to determine the amount of work involved in a chemical or physical change in the chapter on thermodynamics.
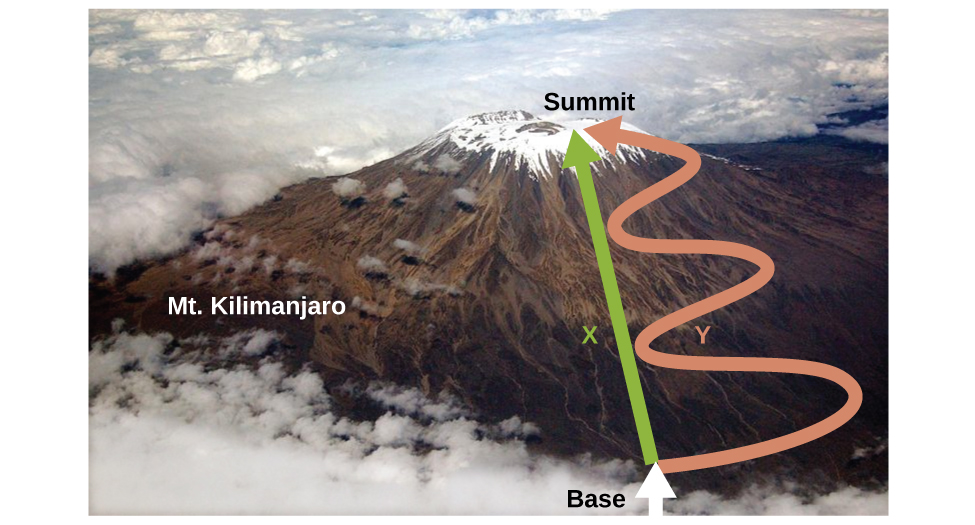
As discussed, the relationship between internal energy, heat, and work can be represented as ΔU = q + w. Internal energy is a type of quantity known as a state function (or state variable), whereas heat and work are not state functions. The value of a state function depends only on the state that a system is in, and not on how that state is reached. If a quantity is not a state function, then its value does depend on how the state is reached. An example of a state function is altitude or elevation. If you stand on the summit of Mt. Kilimanjaro, you are at an altitude of 5895 m, and it does not matter whether you hiked there or parachuted there. The distance you traveled to the top of Kilimanjaro, however, is not a state function. You could climb to the summit by a direct route or by a more roundabout, circuitous path (Figure \(\PageIndex{2}\)). The distances traveled would differ (distance is not a state function) but the elevation reached would be the same (altitude is a state function).
Chemists ordinarily use a property known as enthalpy (\(H\)) to describe the thermodynamics of chemical and physical processes. Enthalpy is defined as the sum of a system’s internal energy (\(U\)) and the mathematical product of its pressure (\(P\)) and volume (\(V\)):
\[H=U+PV \label{5.4.2} \]
Since it is derived from three state functions (\(U\), \(P\), and \(V\)), enthalpy is also a state function. Enthalpy values for specific substances cannot be measured directly; only enthalpy changes for chemical or physical processes can be determined. For processes that take place at constant pressure (a common condition for many chemical and physical changes), the enthalpy change (\(ΔH\)) is:
\[ΔH=ΔU+PΔV\label{5.4.3} \]
The mathematical product \(PΔV\) represents work (\(w\)), namely, expansion or pressure-volume work as noted. By their definitions, the arithmetic signs of ΔV and w will always be opposite:
\[PΔV=−w \label{5.4.4} \]
Substituting Equation \ref{5.4.4} and the definition of internal energy (Equation \ref{5.4.1}) into Equation \ref{5.4.3} yields:
\[ \begin{align} ΔH&=ΔU+PΔV \\[4pt] &=q_\ce{p}+\cancel{w}−\cancel{w} \\[4pt] &=q_\ce{p} \label{5.4.5} \end{align} \]
where \(q_p\) is the heat of reaction under conditions of constant pressure.
And so, if a chemical or physical process is carried out at constant pressure with the only work done caused by expansion or contraction, then the heat flow (\(q_\ce{p}\)) and enthalpy change (\(ΔH\)) for the process are equal.
The heat given off when you operate a Bunsen burner is equal to the enthalpy change of the methane combustion reaction that takes place, since it occurs at the essentially constant pressure of the atmosphere. On the other hand, the heat produced by a reaction measured in a bomb calorimeter is not equal to \(ΔH\) because the closed, constant-volume metal container prevents expansion work from occurring. Chemists usually perform experiments under normal atmospheric conditions, at constant external pressure with \(q = ΔH\), which makes enthalpy the most convenient choice for determining heat.
The following conventions apply when we use \(ΔH\):
- Chemists use a thermochemical equation to represent the changes in both matter and energy. In a thermochemical equation, the enthalpy change of a reaction is shown as a ΔH value following the equation for the reaction. This \(ΔH\) value indicates the amount of heat associated with the reaction involving the number of moles of reactants and products as shown in the chemical equation. For example, consider this equation: \[\ce{H2(g) + 1/2 O2(g) ⟶ H2O (l)} \;\; ΔH=\mathrm{−286\:kJ} \label{5.4.6} \] This equation indicates that when 1 mole of hydrogen gas and 12 mole of oxygen gas at some temperature and pressure change to 1 mole of liquid water at the same temperature and pressure, 286 kJ of heat are released to the surroundings. If the coefficients of the chemical equation are multiplied by some factor, the enthalpy change must be multiplied by that same factor (ΔH is an extensive property).
\[\begin {align*} &\textrm{(two-fold increase in amounts)}\label{5.4.7}\\ &\ce{2H2}(g)+\ce{O2}(g)⟶\ce{2H2O}(l)\hspace{20px}ΔH=\mathrm{2×(−286\:kJ)=−572\:kJ}\\ &\textrm{(two-fold decrease in amounts)}\\ &\frac{1}{2}\ce{H2}(g)+\dfrac{1}{4}\ce{O2}(g)⟶\frac{1}{2}\ce{H2O}(l)\hspace{20px}ΔH=\mathrm{\frac{1}{2}×(−286\:kJ)=−143\:kJ} \end {align*} \label{5.4.6B} \]
- The enthalpy change of a reaction depends on the physical state of the reactants and products of the reaction (whether we have gases, liquids, solids, or aqueous solutions), so these must be shown. For example, when 1 mole of hydrogen gas and 12 mole of oxygen gas change to 1 mole of liquid water at the same temperature and pressure, 286 kJ of heat are released. If gaseous water forms, only 242 kJ of heat are released.
\[\ce{ H2(g) + 1/2 O2(g) ⟶ H2O(g)} \;\;\; ΔH=\ce{−242\:kJ} \label{5.4.7B} \]
- A negative value of an enthalpy change, ΔH, indicates an exothermic reaction; a positive value of ΔH indicates an endothermic reaction. If the direction of a chemical equation is reversed, the arithmetic sign of its ΔH is changed (a process that is endothermic in one direction is exothermic in the opposite direction).
When 0.0500 mol of HCl(aq) reacts with 0.0500 mol of NaOH(aq) to form 0.0500 mol of NaCl(aq), 2.9 kJ of heat are produced. What is ΔH, the enthalpy change, per mole of acid reacting, for the acid-base reaction run under the conditions described ?
\[\ce{HCl (aq) + NaOH(aq) \rightarrow NaCl (aq) + H2O(l)} \nonumber \]
Solution
For the reaction of 0.0500 mol acid (HCl), q = −2.9 kJ. This ratio
\[\mathrm{\dfrac{−2.9 \; kJ}{0.0500\; mol\; HCl}} \nonumber \]
can be used as a conversion factor to find the heat produced when 1 mole of HCl reacts:
\[ΔH =\mathrm{1\; \cancel{mol\; HCl} \times \dfrac{ −2.9\; kJ}{0.0500 \;\cancel{ mol\; HCl}} =−58\; kJ} \nonumber \]
The enthalpy change when 1 mole of HCl reacts is −58 kJ. Since that is the number of moles in the chemical equation, we write the thermochemical equation as:
\[\ce{HCl}_{(aq)}+\ce{NaOH}_{(aq)}⟶\ce{NaCl}_{(aq)}+\ce{H_2O}_{(l)} \;\;\; ΔH=\mathrm{−58\;kJ} \nonumber \]
When 1.34 g Zn(s) reacts with 60.0 mL of 0.750 M HCl(aq), 3.14 kJ of heat are produced. Determine the enthalpy change per mole of zinc reacting for the reaction:
\[ \ce{Zn}_{(s)}+\ce{2HCl}_{(aq)}⟶\ce{ZnCl}_{(aq)}+\ce{H}_{2(g)} \nonumber \]
- Answer
-
ΔH = −153 kJ
Be sure to take both stoichiometry and limiting reactants into account when determining the ΔH for a chemical reaction.
A gummy bear contains 2.67 g sucrose, C12H22O11. When it reacts with 7.19 g potassium chlorate, KClO3, 43.7 kJ of heat are produced. Determine the enthalpy change for the reaction
\[\ce{C12H22O11}(aq)+\ce{8KClO3}(aq)⟶\ce{12CO2}(g)+\ce{11H2O}(l)+\ce{8KCl}(aq) \nonumber \]
Solution
We have \(\mathrm{2.67\:\cancel{g}×\dfrac{1\:mol}{342.3\:\cancel{g}}=0.00780\:mol\:C_{12}H_{22}O_{11}}\) available, and
\(\mathrm{7.19\:\cancel{g}×\dfrac{1\:mol}{122.5\:\cancel{g}}=0.0587\:mol\:KClO_3}\) available.
Since
\(\mathrm{0.0587\:mol\:KClO_3×\dfrac{1\:mol\:\ce{C12H22O11}}{8\:mol\:KClO_3}=0.00734\:mol\:\ce{C12H22O11}}\)
is needed, C12H22O11 is the excess reactant and KClO3 is the limiting reactant.
The reaction uses 8 mol KClO3, and the conversion factor is \(\mathrm{\dfrac{−43.7\:kJ}{0.0587\:mol\:KClO_3}}\), so we have \(ΔH=\mathrm{8\:mol×\dfrac{−43.7\:kJ}{0.0587\:mol\:KClO_3}=−5960\:kJ}\). The enthalpy change for this reaction is −5960 kJ, and the thermochemical equation is:
\[\ce{C12H22O11 + 8KClO3⟶12CO2 + 11H2O + 8KCl}\hspace{20px}ΔH=\ce{−5960\:kJ} \nonumber \]
When 1.42 g of iron reacts with 1.80 g of chlorine, 3.22 g of \(\ce{FeCl}_{2(s)}\) and 8.60 kJ of heat is produced. What is the enthalpy change for the reaction when 1 mole of \(\ce{FeCl2(s)}\) is produced?
- Answer
-
ΔH = −338 kJ
Enthalpy changes are typically tabulated for reactions in which both the reactants and products are at the same conditions. A standard state is a commonly accepted set of conditions used as a reference point for the determination of properties under other different conditions. For chemists, the IUPAC standard state refers to materials under a pressure of 1 bar and solutions at 1 M, and does not specify a temperature (it used too). Many thermochemical tables list values with a standard state of 1 atm. Because the ΔH of a reaction changes very little with such small changes in pressure (1 bar = 0.987 atm), ΔH values (except for the most precisely measured values) are essentially the same under both sets of standard conditions. We will include a superscripted “o” in the enthalpy change symbol to designate standard state. Since the usual (but not technically standard) temperature is 298.15 K, we will use a subscripted “298” to designate this temperature. Thus, the symbol (\(ΔH^\circ_{298}\)) is used to indicate an enthalpy change for a process occurring under these conditions. (The symbol ΔH is used to indicate an enthalpy change for a reaction occurring under nonstandard conditions.)
The enthalpy changes for many types of chemical and physical processes are available in the reference literature, including those for combustion reactions, phase transitions, and formation reactions. As we discuss these quantities, it is important to pay attention to the extensive nature of enthalpy and enthalpy changes. Since the enthalpy change for a given reaction is proportional to the amounts of substances involved, it may be reported on that basis (i.e., as the ΔH for specific amounts of reactants). However, we often find it more useful to divide one extensive property (ΔH) by another (amount of substance), and report a per-amount intensive value of ΔH, often “normalized” to a per-mole basis. (Note that this is similar to determining the intensive property specific heat from the extensive property heat capacity, as seen previously.)
Enthalpy of Combustion
Standard enthalpy of combustion (\(ΔH_C^\circ\)) is the enthalpy change when 1 mole of a substance burns (combines vigorously with oxygen) under standard state conditions; it is sometimes called “heat of combustion.” For example, the enthalpy of combustion of ethanol, −1366.8 kJ/mol, is the amount of heat produced when one mole of ethanol undergoes complete combustion at 25 °C and 1 atmosphere pressure, yielding products also at 25 °C and 1 atm.
\[\ce{C2H5OH}(l)+\ce{3O2}(g)⟶\ce{2CO2}+\ce{3H2O}(l)\hspace{20px}ΔH_{298}^\circ=\mathrm{−1366.8\: kJ} \label{5.4.8} \]
Enthalpies of combustion for many substances have been measured; a few of these are listed in Table \(\PageIndex{1}\). Many readily available substances with large enthalpies of combustion are used as fuels, including hydrogen, carbon (as coal or charcoal), and hydrocarbons (compounds containing only hydrogen and carbon), such as methane, propane, and the major components of gasoline.
| Substance | Combustion Reaction | Enthalpy of Combustion \(ΔH_c^\circ \left(\mathrm{\dfrac{kJ}{mol} \:at\:25°C}\right)\) |
|---|---|---|
| carbon | \(\ce{C}(s)+\ce{O2}(g)⟶\ce{CO2}(g)\) | −393.5 |
| hydrogen | \(\ce{H2}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{H2O}(l)\) | −285.8 |
| magnesium | \(\ce{Mg}(s)+\frac{1}{2}\ce{O2}(g)⟶\ce{MgO}(s)\) | −601.6 |
| sulfur | \(\ce{S}(s)+\ce{O2}(g)⟶\ce{SO2}(g)\) | −296.8 |
| carbon monoxide | \(\ce{CO}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO2}(g)\) | −283.0 |
| methane | \(\ce{CH4}(g)+\ce{2O2}(g)⟶\ce{CO2}(g)+\ce{2H2O}(l)\) | −890.8 |
| acetylene | \(\ce{C2H2}(g)+\dfrac{5}{2}\ce{O2}(g)⟶\ce{2CO2}(g)+\ce{H2O}(l)\) | −1301.1 |
| ethanol | \(\ce{C2H5OH}(l)+\ce{3O2}(g)⟶\ce{CO2}(g)+\ce{3H2O}(l)\) | −1366.8 |
| methanol | \(\ce{CH3OH}(l)+\dfrac{3}{2}\ce{O2}(g)⟶\ce{CO2}(g)+\ce{2H2O}(l)\) | −726.1 |
| isooctane | \(\ce{C8H18}(l)+\dfrac{25}{2}\ce{O2}(g)⟶\ce{8CO2}(g)+\ce{9H2O}(l)\) | −5461 |
As Figure \(\PageIndex{3}\) suggests, the combustion of gasoline is a highly exothermic process. Let us determine the approximate amount of heat produced by burning 1.00 L of gasoline, assuming the enthalpy of combustion of gasoline is the same as that of isooctane, a common component of gasoline. The density of isooctane is 0.692 g/mL.

Solution
Starting with a known amount (1.00 L of isooctane), we can perform conversions between units until we arrive at the desired amount of heat or energy. The enthalpy of combustion of isooctane provides one of the necessary conversions. Table \(\PageIndex{1}\) gives this value as −5460 kJ per 1 mole of isooctane (C8H18).
Using these data,
\[\mathrm{1.00\:\cancel{L\:\ce{C8H18}}×\dfrac{1000\:\cancel{mL\:\ce{C8H18}}}{1\:\cancel{L\:\ce{C8H18}}}×\dfrac{0.692\:\cancel{g\:\ce{C8H18}}}{1\:\cancel{mL\:\ce{C8H18}}}×\dfrac{1\:\cancel{mol\:\ce{C8H18}}}{114\:\cancel{g\:\ce{C8H18}}}×\dfrac{−5460\:kJ}{1\:\cancel{mol\:\ce{C8H18}}}=−3.31×10^4\:kJ} \nonumber \]
The combustion of 1.00 L of isooctane produces 33,100 kJ of heat. (This amount of energy is enough to melt 99.2 kg, or about 218 lbs, of ice.)
Note: If you do this calculation one step at a time, you would find:
\(\begin {align*}
&\mathrm{1.00\:L\:\ce{C8H18}⟶1.00×10^3\:mL\:\ce{C8H18}}\\
&\mathrm{1.00×10^3\:mL\:\ce{C8H18}⟶692\:g\:\ce{C8H18}}\\
&\mathrm{692\:g\:\ce{C8H18}⟶6.07\:mol\:\ce{C8H18}}\\
&\mathrm{692\:g\:\ce{C8H18}⟶−3.31×10^4\:kJ}
\end {align*}\)
How much heat is produced by the combustion of 125 g of acetylene?
- Answer
-
6.25 × 103 kJ
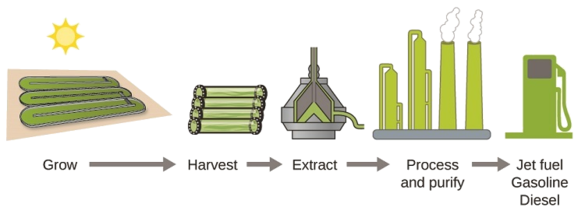
As reserves of fossil fuels diminish and become more costly to extract, the search is ongoing for replacement fuel sources for the future. Among the most promising biofuels are those derived from algae (Figure \(\PageIndex{4}\)). The species of algae used are nontoxic, biodegradable, and among the world’s fastest growing organisms. About 50% of algal weight is oil, which can be readily converted into fuel such as biodiesel. Algae can yield 26,000 gallons of biofuel per hectare—much more energy per acre than other crops. Some strains of algae can flourish in brackish water that is not usable for growing other crops. Algae can produce biodiesel, biogasoline, ethanol, butanol, methane, and even jet fuel.

According to the US Department of Energy, only 39,000 square kilometers (about 0.4% of the land mass of the US or less than \(\dfrac{1}{7}\) of the area used to grow corn) can produce enough algal fuel to replace all the petroleum-based fuel used in the US. The cost of algal fuels is becoming more competitive—for instance, the US Air Force is producing jet fuel from algae at a total cost of under $5 per gallon. The process used to produce algal fuel is as follows: grow the algae (which use sunlight as their energy source and CO2 as a raw material); harvest the algae; extract the fuel compounds (or precursor compounds); process as necessary (e.g., perform a transesterification reaction to make biodiesel); purify; and distribute (Figure \(\PageIndex{5}\)).
Standard Enthalpy of Formation
A standard enthalpy of formation \(ΔH^\circ_\ce{f}\) is an enthalpy change for a reaction in which exactly 1 mole of a pure substance is formed from free elements in their most stable states under standard state conditions. These values are especially useful for computing or predicting enthalpy changes for chemical reactions that are impractical or dangerous to carry out, or for processes for which it is difficult to make measurements. If we have values for the appropriate standard enthalpies of formation, we can determine the enthalpy change for any reaction, which we will practice in the next section on Hess’s law.
The standard enthalpy of formation of CO2(g) is −393.5 kJ/mol. This is the enthalpy change for the exothermic reaction:
\[\ce{C}(s)+\ce{O2}(g)⟶\ce{CO2}(g)\hspace{20px}ΔH^\circ_\ce{f}=ΔH^\circ_{298}=−393.5\:\ce{kJ} \label{5.4.9} \]
starting with the reactants at a pressure of 1 atm and 25 °C (with the carbon present as graphite, the most stable form of carbon under these conditions) and ending with one mole of CO2, also at 1 atm and 25 °C. For nitrogen dioxide, \(\ce{NO}_{2(g)}\), \(ΔH^\circ_\ce{f}\) is 33.2 kJ/mol. This is the enthalpy change for the reaction:
\[\frac{1}{2}\ce{N2}(g)+\ce{O2}(g)⟶\ce{NO2}(g)\hspace{20px}ΔH^\circ_\ce{f}=ΔH^\circ_{298}=+33.2\: \ce{kJ} \label{5.4.10} \]
A reaction equation with \(\frac{1}{2}\) mole of N2 and 1 mole of O2 is correct in this case because the standard enthalpy of formation always refers to 1 mole of product, NO2(g).
You will find a table of standard enthalpies of formation of many common substances in Tables T1 and T2. These values indicate that formation reactions range from highly exothermic (such as −2984 kJ/mol for the formation of P4O10) to strongly endothermic (such as +226.7 kJ/mol for the formation of acetylene, C2H2). By definition, the standard enthalpy of formation of an element in its most stable form is equal to zero under standard conditions, which is 1 atm for gases and 1 M for solutions.
Ozone, O3(g), forms from oxygen, O2(g), by an endothermic process. Ultraviolet radiation is the source of the energy that drives this reaction in the upper atmosphere. Assuming that both the reactants and products of the reaction are in their standard states, determine the standard enthalpy of formation, \(ΔH^\circ_\ce{f}\) of ozone from the following information:
\[\ce{3O2}(g)⟶\ce{2O3}(g)\hspace{20px}ΔH^\circ_{298}=+286\: \ce{kJ} \nonumber \]
Solution \(ΔH^\circ_\ce{f}\) is the enthalpy change for the formation of one mole of a substance in its standard state from the elements in their standard states. Thus, \(ΔH^\circ_\ce{f}\) for O3(g) is the enthalpy change for the reaction:
\[\dfrac{3}{2}\ce{O2}(g)⟶\ce{O3}(g) \nonumber \]
For the formation of 2 mol of O3(g), \(ΔH^\circ_{298}=+286\: \ce{kJ}\). This ratio, \(\mathrm{\left(\dfrac{286\:kJ}{2\:mol\:O_3}\right)}\), can be used as a conversion factor to find the heat produced when 1 mole of O3(g) is formed, which is the enthalpy of formation for O3(g):
\[ΔH^\circ \ce{\:for\:1\:mole\: of\:O_3}(g)=\mathrm{1\:\cancel{mol\:O_3}×\dfrac{286\:kJ}{2\:\cancel{mol\:O_3}}=143\:kJ} \nonumber \]
Therefore, \(ΔH^\circ_\ce{f}[\ce{O3}(g)]=\ce{+143\: kJ/mol}\).
Hydrogen gas, H2, reacts explosively with gaseous chlorine, Cl2, to form hydrogen chloride, HCl(g). What is the enthalpy change for the reaction of 1 mole of H2(g) with 1 mole of Cl2(g) if both the reactants and products are at standard state conditions? The standard enthalpy of formation of HCl(g) is −92.3 kJ/mol.
- Answer
-
For the reaction
\[\ce{H2}(g)+\ce{Cl2}(g)⟶\ce{2HCl}(g)\hspace{20px}ΔH^\circ_{298}=\mathrm{−184.6\:kJ} \nonumber \]
Write the heat of formation reaction equations for:
- \(\ce{C2H_5OH}_{(l)}\)
- \(\ce{Ca_3(PO_4)}_{2(s)}\)
Solution
Remembering that \(ΔH^\circ_\ce{f}\) reaction equations are for forming 1 mole of the compound from its constituent elements under standard conditions, we have:
- \(\ce{2C}(s,\:\ce{graphite})+\ce{3H2}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{C2H5OH}(l)\)
- \(\ce{3Ca}(s)+\frac{1}{2}\ce{P4}(s)+\ce{4O2}(g)⟶\ce{Ca3(PO4)2}(s)\)
Note: The standard state of carbon is graphite, and phosphorus exists as \(P_4\).
Write the heat of formation reaction equations for:
- \(\ce{C_2H_5OC_2H}_{5(l)}\)
- \(\ce{Na_2CO}_{3(s)}\)
- Answer a
-
\(\ce{4C}(s,\:\ce{graphite})+\ce{5H2}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{C2H5OC2H5}(l)\);
- Answer b
-
\(\ce{2Na}(s)+\ce{C}(s,\:\ce{graphite})+\dfrac{3}{2}\ce{O2}(g)⟶\ce{Na2CO3}(s)\)
Hess’s Law
There are two ways to determine the amount of heat involved in a chemical change: measure it experimentally, or calculate it from other experimentally determined enthalpy changes. Some reactions are difficult, if not impossible, to investigate and make accurate measurements for experimentally. And even when a reaction is not hard to perform or measure, it is convenient to be able to determine the heat involved in a reaction without having to perform an experiment.
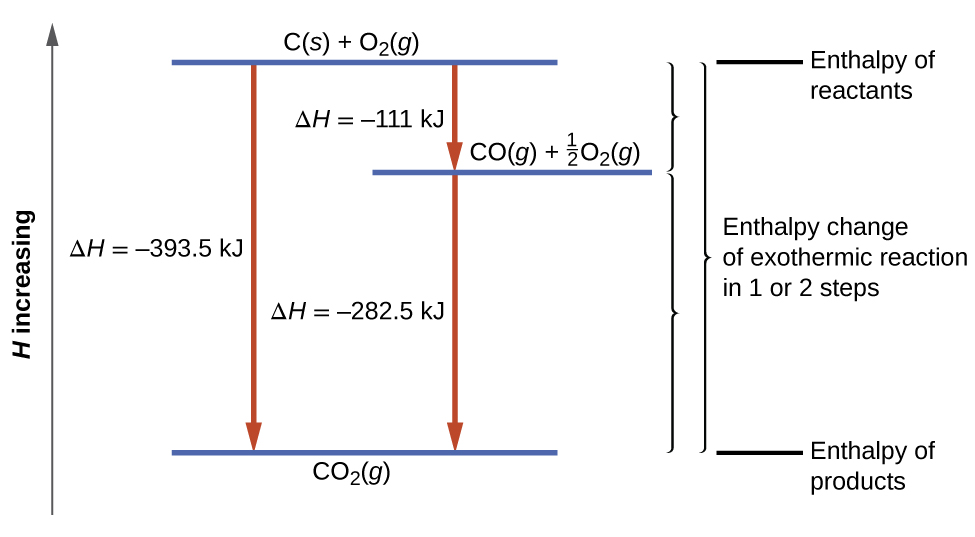
This type of calculation usually involves the use of Hess’s law, which states: If a process can be written as the sum of several stepwise processes, the enthalpy change of the total process equals the sum of the enthalpy changes of the various steps. Hess’s law is valid because enthalpy is a state function: Enthalpy changes depend only on where a chemical process starts and ends, but not on the path it takes from start to finish. For example, we can think of the reaction of carbon with oxygen to form carbon dioxide as occurring either directly or by a two-step process. The direct process is written:
\[\ce{C}_{(s)}+\ce{O}_{2(g)}⟶\ce{CO}_{2(g)}\;\;\;ΔH^∘_{298}=\mathrm{−394\;kJ} \label{ 5.4.11} \]
In the two-step process, first carbon monoxide is formed:
\[\ce{C}_{(s)}+\dfrac{1}{2}\ce{O}_{2(g)}⟶\ce{CO}_{(g)}\;\;\;ΔH^∘_{298}=\mathrm{−111\;kJ} \label{ 5.4.12} \]
Then, carbon monoxide reacts further to form carbon dioxide:
\[\ce{CO} {(g)}+\dfrac{1}{2}\ce{O2}(g)⟶\ce{CO}_2 {(g)}\;\;\;ΔH^∘_{298}=\mathrm{−283\;kJ} \label{ 5.4.13} \]
The equation describing the overall reaction is the sum of these two chemical changes:
\[\begin {align*} &\textrm{Step 1:} \:\ce{C}(s)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO}(g)\\ &\underline{\textrm{Step 2:} \:\ce{CO}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO2}(g)}\\ &\textrm{Sum:} \:\ce{C}(s)+\frac{1}{2}\ce{O2}(g)+\ce{CO}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO}(g)+\ce{CO2}(g) \end {align*} \label{5.4.14} \]
Because the CO produced in Step 1 is consumed in Step 2, the net change is:
\[\ce{C}_{(s)}+\ce{O}_{2(g)}⟶\ce{CO}_{2(g)} \label{5.4.15} \]
According to Hess’s law, the enthalpy change of the reaction will equal the sum of the enthalpy changes of the steps. We can apply the data from the experimental enthalpies of combustion in Table \(\PageIndex{1}\) to find the enthalpy change of the entire reaction from its two steps:
\[\begin {align*}
&\ce{C}(s)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO}(g)&&ΔH^\circ_{298}=\mathrm{−111\:kJ}\\
&\ce{CO}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{CO2}(g)&&ΔH^\circ_{298}=\mathrm{−283\:kJ}\\
&\overline{\ce{C}(s)+\ce{O2}(g)⟶\ce{CO2}(g)\hspace{25px}}&&\overline{ΔH^\circ_{298}=\mathrm{−394\:kJ}}
\end {align*} \label{5.4.16} \]
The result is shown in Figure \(\PageIndex{6}\). We see that ΔH of the overall reaction is the same whether it occurs in one step or two. This finding (overall ΔH for the reaction = sum of ΔH values for reaction “steps” in the overall reaction) is true in general for chemical and physical processes.
Before we further practice using Hess’s law, let us recall two important features of ΔH.
- ΔH is directly proportional to the quantities of reactants or products. For example, the enthalpy change for the reaction forming 1 mole of NO2(g) is +33.2 kJ: \[\frac{1}{2}\ce{N2}(g)+\ce{O2}(g)⟶\ce{NO2}(g)\hspace{20px}ΔH=\mathrm{+33.2\: kJ} \label{5.4.17} \]
When 2 moles of NO2 (twice as much) are formed, the ΔH will be twice as large: \[\ce{N2}(g)+\ce{2O2}(g)⟶\ce{2NO2}(g)\hspace{20px}ΔH=\mathrm{+66.4\: kJ} \label{5.4.18} \]
In general, if we multiply or divide an equation by a number, then the enthalpy change should also be multiplied or divided by the same number.
- ΔH for a reaction in one direction is equal in magnitude and opposite in sign to ΔH for the reaction in the reverse direction. For example, given that: \[\ce{H2}(g)+\ce{Cl2}(g)⟶\ce{2HCl}(g)\hspace{20px}ΔH=\mathrm{−184.6\:kJ} \label{5.4.19} \]
Then, for the “reverse” reaction, the enthalpy change is also “reversed”: \[\ce{2HCl}(g)⟶\ce{H2}(g)+\ce{Cl2}(g)\hspace{20px}ΔH=\mathrm{+184.6\: kJ} \label{5.4.20} \]
Using Hess’s Law Determine the enthalpy of formation, \(ΔH^\circ_\ce{f}\), of FeCl3(s) from the enthalpy changes of the following two-step process that occurs under standard state conditions:
\[\ce{Fe}(s)+\ce{Cl2}(g)⟶\ce{FeCl2}(s)\hspace{20px}ΔH°=\mathrm{−341.8\:kJ} \nonumber \]
\[\ce{FeCl2}(s)+\frac{1}{2}\ce{Cl2}(g)⟶\ce{FeCl3}(s)\hspace{20px}ΔH°=\mathrm \nonumber{−57.7\:kJ} \nonumber \]
Solution
We are trying to find the standard enthalpy of formation of FeCl3(s), which is equal to ΔH° for the reaction:
\[\ce{Fe}(s)+\frac{3}{2}\ce{Cl2}(g)⟶\ce{FeCl3}(s)\hspace{20px}ΔH^\circ_\ce{f}=\:? \nonumber \]
Looking at the reactions, we see that the reaction for which we want to find ΔH° is the sum of the two reactions with known ΔH values, so we must sum their ΔHs:
\[\ce{Fe}(s)+\ce{Cl2}(g)⟶\ce{FeCl2}(s)\hspace{59px}ΔH°=\mathrm{−341.8\:kJ}\\ \underline{\ce{FeCl2}(s)+\frac{1}{2}\ce{Cl2}(g)⟶\ce{FeCl3}(s)\hspace{20px}ΔH°=\mathrm{−57.7\:kJ}}\\ \ce{Fe}(s)+\frac{1}{2}\ce{Cl2}(g)⟶\ce{FeCl3}(s)\hspace{43px}ΔH°=\mathrm{−399.5\:kJ} \nonumber \]
The enthalpy of formation, \(ΔH^\circ_\ce{f}\), of FeCl3(s) is −399.5 kJ/mol.
Calculate ΔH for the process:
\[\ce{N2}(g)+\ce{2O2}(g)⟶\ce{2NO2}(g) \nonumber \]
from the following information:
\[\ce{N2}(g)+\ce{O2}(g)⟶\ce{2NO}(g)\hspace{20px}ΔH=\mathrm{180.5\:kJ} \nonumber \]
\[\ce{NO}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{NO2}(g)\hspace{20px}ΔH=\mathrm{−57.06\:kJ} \nonumber \]
- Answer
-
66.4 kJ
Here is a less straightforward example that illustrates the thought process involved in solving many Hess’s law problems. It shows how we can find many standard enthalpies of formation (and other values of ΔH) if they are difficult to determine experimentally.
Using Hess’s Law Chlorine monofluoride can react with fluorine to form chlorine trifluoride:
(i) \(\ce{ClF}(g)+\ce{F2}(g)⟶\ce{ClF3}(g)\hspace{20px}ΔH°=\:?\)
Use the reactions here to determine the ΔH° for reaction (i):
(ii) \(\ce{2OF2}(g)⟶\ce{O2}(g)+\ce{2F2}(g)\hspace{20px}ΔH^\circ_{(ii)}=\mathrm{−49.4\:kJ}\)
(iii) \(\ce{2ClF}(g)+\ce{O2}(g)⟶\ce{Cl2O}(g)+\ce{OF2}(g)\hspace{20px}ΔH^\circ_{(iii)}=\mathrm{+205.6\: kJ}\)
(iv) \(\ce{ClF3}(g)+\ce{O2}(g)⟶\frac{1}{2}\ce{Cl2O}(g)+\dfrac{3}{2}\ce{OF2}(g)\hspace{20px}ΔH^\circ_{(iv)}=\mathrm{+266.7\: kJ}\)
Solution
Our goal is to manipulate and combine reactions (ii), (iii), and (iv) such that they add up to reaction (i). Going from left to right in (i), we first see that \(\ce{ClF}_{(g)}\) is needed as a reactant. This can be obtained by multiplying reaction (iii) by \(\frac{1}{2}\), which means that the ΔH° change is also multiplied by \(\frac{1}{2}\):
\[\ce{ClF}(g)+\frac{1}{2}\ce{O2}(g)⟶\frac{1}{2}\ce{Cl2O}(g)+\frac{1}{2}\ce{OF2}(g)\hspace{20px} ΔH°=\frac{1}{2}(205.6)=+102.8\: \ce{kJ} \nonumber \]
Next, we see that \(\ce{F_2}\) is also needed as a reactant. To get this, reverse and halve reaction (ii), which means that the ΔH° changes sign and is halved:
\[\frac{1}{2}\ce{O2}(g)+\ce{F2}(g)⟶\ce{OF2}(g)\hspace{20px}ΔH°=+24.7\: \ce{kJ} \nonumber \]
To get ClF3 as a product, reverse (iv), changing the sign of ΔH°:
\[\frac{1}{2}\ce{Cl2O}(g)+\dfrac{3}{2}\ce{OF2}(g)⟶\ce{ClF3}(g)+\ce{O2}(g)\hspace{20px}ΔH°=\mathrm{−266.7\: kJ} \nonumber \]
Now check to make sure that these reactions add up to the reaction we want:
\[\begin {align*}
&\ce{ClF}(g)+\frac{1}{2}\ce{O2}(g)⟶\frac{1}{2}\ce{Cl2O}(g)+\frac{1}{2}\ce{OF2}(g)&&ΔH°=\mathrm{+102.8\: kJ}\\
&\frac{1}{2}\ce{O2}(g)+\ce{F2}(g)⟶\ce{OF2}(g)&&ΔH°=\mathrm{+24.7\: kJ}\\
&\frac{1}{2}\ce{Cl2O}(g)+\dfrac{3}{2}\ce{OF2}(g)⟶\ce{ClF3}(g)+\ce{O2}(g)&&ΔH°=\mathrm{−266.7\:kJ}\\
&\overline{\ce{ClF}(g)+\ce{F2}⟶\ce{ClF3}(g)\hspace{130px}}&&\overline{ΔH°=\mathrm{−139.2\:kJ}}
\end {align*} \nonumber \]
Reactants \(\frac{1}{2}\ce{O2}\) and \(\frac{1}{2}\ce{O2}\) cancel out product O2; product \(\frac{1}{2}\ce{Cl2O}\) cancels reactant \(\frac{1}{2}\ce{Cl2O}\); and reactant \(\dfrac{3}{2}\ce{OF2}\) is cancelled by products \(\frac{1}{2}\ce{OF2}\) and OF2. This leaves only reactants ClF(g) and F2(g) and product ClF3(g), which are what we want. Since summing these three modified reactions yields the reaction of interest, summing the three modified ΔH° values will give the desired ΔH°:
Aluminum chloride can be formed from its elements:
(i) \(\ce{2Al}(s)+\ce{3Cl2}(g)⟶\ce{2AlCl3}(s)\hspace{20px}ΔH°=\:?\)
Use the reactions here to determine the ΔH° for reaction (i):
(ii) \(\ce{HCl}(g)⟶\ce{HCl}(aq)\hspace{20px}ΔH^\circ_{(ii)}=\mathrm{−74.8\:kJ}\)
(iii) \(\ce{H2}(g)+\ce{Cl2}(g)⟶\ce{2HCl}(g)\hspace{20px}ΔH^\circ_{(iii)}=\mathrm{−185\:kJ}\)
(iv) \(\ce{AlCl3}(aq)⟶\ce{AlCl3}(s)\hspace{20px}ΔH^\circ_{(iv)}=\mathrm{+323\:kJ/mol}\)
(v) \(\ce{2Al}(s)+\ce{6HCl}(aq)⟶\ce{2AlCl3}(aq)+\ce{3H2}(g)\hspace{20px}ΔH^\circ_{(v)}=\mathrm{−1049\:kJ}\)
- Answer
-
−1407 kJ
We also can use Hess’s law to determine the enthalpy change of any reaction if the corresponding enthalpies of formation of the reactants and products are available. The stepwise reactions we consider are: (i) decompositions of the reactants into their component elements (for which the enthalpy changes are proportional to the negative of the enthalpies of formation of the reactants), followed by (ii) re-combinations of the elements to give the products (with the enthalpy changes proportional to the enthalpies of formation of the products). The standard enthalpy change of the overall reaction is therefore equal to: (ii) the sum of the standard enthalpies of formation of all the products plus (i) the sum of the negatives of the standard enthalpies of formation of the reactants. This is usually rearranged slightly to be written as follows, with \(\sum\) representing “the sum of” and n standing for the stoichiometric coefficients:
\[ΔH^\circ_\ce{reaction}=\sum n×ΔH^\circ_\ce{f}\ce{(products)}−\sum n×ΔH^\circ_\ce{f}\ce{(reactants)} \label{5.4.20B} \]
The following example shows in detail why this equation is valid, and how to use it to calculate the enthalpy change for a reaction of interest.
What is the standard enthalpy change for the reaction:
\[\ce{3NO2}(g)+\ce{H2O}(l)⟶\ce{2HNO3}(aq)+\ce{NO}(g)\hspace{20px}ΔH°=\:? \nonumber \]
Solution 1: Using the Equation
Alternatively, we could use the special form of Hess’s law given previously:
\[ΔH^\circ_\ce{reaction}=∑n×ΔH^\circ_\ce{f}\ce{(products)}−∑n×ΔH^\circ_\ce{f}\ce{(reactants)} \nonumber \]
\[\begin {align*} &=\mathrm{\left[2\:\cancel{mol\:HNO_3}×\dfrac{−207.4\:kJ}{\cancel{mol\:HNO_3\:(\mathit{aq})}}+1\:\cancel{mol\: NO\:(\mathit{g})}×\dfrac{+90.2\: kJ}{\cancel{mol\: NO\:(\mathit{g})}}\right]}\\ &\mathrm{\:−\,\left[3\:\cancel{mol\:NO_2(\mathit{g})}×\dfrac{+33.2\: kJ}{\cancel{mol\:NO_2\:(\mathit{g})}}+1\:\cancel{mol\:H_2O\:(\mathit{l})}×\dfrac{−285.8\:kJ}{\cancel{mol\:H_2O\:(\mathit{l})}}\right]}\\ &=\mathrm{2(−207.4\:kJ)+1(+90.2\: kJ)−3(+33.2\: kJ)−1(−285.8\:kJ)}\\ &=\mathrm{−138.4\:kJ}\end {align*} \nonumber \]
Solution 2: Supporting Why the General Equation Is Valid
We can write this reaction as the sum of the decompositions of 3NO2(g) and 1H2O(l) into their constituent elements, and the formation of 2 HNO3(aq) and 1 NO(g) from their constituent elements. Writing out these reactions, and noting their relationships to the \(ΔH^\circ_\ce{f}\) values for these compounds (from Tables T1 and T2 ), we have:
\[\ce{3NO2}(g)⟶ \dfrac{3}{2} \ce{N2}(g)+ 3 \ce{O2}(g)\hspace{20px}ΔH^\circ_{1}=\mathrm{−99.6\:kJ} \nonumber \]
\[\ce{H2O}(l)⟶\ce{H2}(g)+\frac{1}{2}\ce{O2}(g)\hspace{20px}ΔH^\circ_{2}=+285.8\: \ce{kJ}\:[−1×ΔH^\circ_\ce{f}(\ce{H2O})] \nonumber \]
\[\ce{H2}(g)+\ce{N2}(g)+ 3 \ce{O2}(g)⟶\ce{2HNO3}(aq)\hspace{20px}ΔH^\circ_{3}=−414.8\:kJ\:[2×ΔH^\circ_\ce{f}(\ce{HNO3 \nonumber})] \nonumber \]
\[\frac{1}{2}\ce{N2}(g)+\frac{1}{2}\ce{O2}(g)⟶\ce{NO}(g)\hspace{20px}ΔH^\circ \nonumber_{4}=+90.2\: \ce{kJ}\:[1×(\ce{NO})] \nonumber \]
Summing these reaction equations gives the reaction we are interested in:
\[\ce{3NO2}(g)+\ce{H2O}(l)⟶\ce{2HNO3}(aq)+\ce{NO}(g) \nonumber \]
Summing their enthalpy changes gives the value we want to determine:
\[\begin {align*} ΔH^\circ_\ce{rxn}&=ΔH^\circ_1+ΔH^\circ_2+ΔH^\circ_3+ΔH^\circ_4=\mathrm{(−99.6\:kJ)+(+285.8\: kJ)+(−414.8\:kJ)+(+90.2\: kJ)}\\ &=\mathrm{−138.4\:kJ} \end {align*} \nonumber \]
So the standard enthalpy change for this reaction is ΔH° = −138.4 kJ.
Note that this result was obtained by:
- multiplying the \(ΔH^\circ_\ce{f}\) of each product by its stoichiometric coefficient and summing those values,
- multiplying the \(ΔH^\circ_\ce{f}\) of each reactant by its stoichiometric coefficient and summing those values, and then
- subtracting the result found in step 2 from the result found in step 1.
This is also the procedure in using the general equation, as shown.
Calculate the heat of combustion of 1 mole of ethanol, C2H5OH(l), when H2O(l) and CO2(g) are formed. Use the following enthalpies of formation: C2H5OH(l), −278 kJ/mol; H2O(l), −286 kJ/mol; and CO2(g), −394 kJ/mol.
- Answer
-
−1368 kJ/mol
Summary
If a chemical change is carried out at constant pressure and the only work done is caused by expansion or contraction, q for the change is called the enthalpy change with the symbol ΔH, or \(ΔH^\circ_{298}\) for reactions occurring under standard state conditions. The value of ΔH for a reaction in one direction is equal in magnitude, but opposite in sign, to ΔH for the reaction in the opposite direction, and ΔH is directly proportional to the quantity of reactants and products. Examples of enthalpy changes include enthalpy of combustion, enthalpy of fusion, enthalpy of vaporization, and standard enthalpy of formation. The standard enthalpy of formation, \(ΔH^\circ_\ce{f}\), is the enthalpy change accompanying the formation of 1 mole of a substance from the elements in their most stable states at 1 bar (standard state). Many of the processes are carried out at 298.15 K. If the enthalpies of formation are available for the reactants and products of a reaction, the enthalpy change can be calculated using Hess’s law: If a process can be written as the sum of several stepwise processes, the enthalpy change of the total process equals the sum of the enthalpy changes of the various steps.
Key Equations
- \(ΔU=q+w\)
- \(ΔH^\circ_\ce{reaction}=∑n×ΔH^\circ_\ce{f}\ce{(products)}−∑n×ΔH^\circ_\ce{f}\ce{(reactants)}\)
Footnotes
- 1 For more on algal fuel, see www.theguardian.com/environme...n-fuel-problem.
Glossary
- chemical thermodynamics
- area of science that deals with the relationships between heat, work, and all forms of energy associated with chemical and physical processes
- enthalpy (H)
- sum of a system’s internal energy and the mathematical product of its pressure and volume
- enthalpy change (ΔH)
- heat released or absorbed by a system under constant pressure during a chemical or physical process
- expansion work (pressure-volume work)
- work done as a system expands or contracts against external pressure
- first law of thermodynamics
- internal energy of a system changes due to heat flow in or out of the system or work done on or by the system
- Hess’s law
- if a process can be represented as the sum of several steps, the enthalpy change of the process equals the sum of the enthalpy changes of the steps
- hydrocarbon
- compound composed only of hydrogen and carbon; the major component of fossil fuels
- internal energy (U)
- total of all possible kinds of energy present in a substance or substances
- standard enthalpy of combustion (\(ΔH^\circ_\ce{c}\))
- heat released when one mole of a compound undergoes complete combustion under standard conditions
- standard enthalpy of formation (\(ΔH^\circ_\ce{f}\))
- enthalpy change of a chemical reaction in which 1 mole of a pure substance is formed from its elements in their most stable states under standard state conditions
- standard state
- set of physical conditions as accepted as common reference conditions for reporting thermodynamic properties; 1 bar of pressure, and solutions at 1 molar concentrations, usually at a temperature of 298.15 K
- state function
- property depending only on the state of a system, and not the path taken to reach that state