
26.6: Applications of Coordination Compounds
- Page ID
- 219366
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Chelating Agents for Poising (EDTA)
Biomolecules (Porphorins and Hemoglogin)
Drugs and Health (cis-Platin)
- To become familiar with some of the roles of transition-metal complexes in biological systems.
In this section, we describe several systems that illustrate the roles transition metals play in biological systems. Our goal is for you to understand why the chemical properties of these elements make them essential for life. We begin with a discussion of the strategies organisms use to extract transition metals from their environment. The section continues with a brief discussion of the use of transition metals in reactions that involve the transfer of electrons, reactions of small molecules such as O2, Lewis-acid catalysis, and the generation of reactive organic radicals.
Uptake and Storage of Transition Metals
There are three possible dietary levels for any essential element: deficient, optimal, and toxic, in order of increasing concentration in the diet. If the concentration of an essential element in the diet is too low, an organism must be able to extract the element from the environment and concentrate it. If the concentration of an essential element in the diet is too high, an organism must be able to limit its intake to avoid toxic effects. Moreover, organisms must be able to switch off the uptake process rapidly if dietary levels rise suddenly, and they must be able to store essential elements for future use.
Three distinct steps are involved in transition metal uptake. First, the metal must be “mobilized” from the environment and brought into contact with a cell in a form that can be absorbed. Second, the metal must be transported across the cell membrane into the cell. Third, the element must be transported to its point of utilization within a cell or to other cells within the organism. In our discussion, we focus on the uptake, transport, and storage of iron, which illustrates the most important points. Because iron deficiency (anemia) is the most widespread nutritional deficiency known in humans, the uptake of iron is especially well understood.
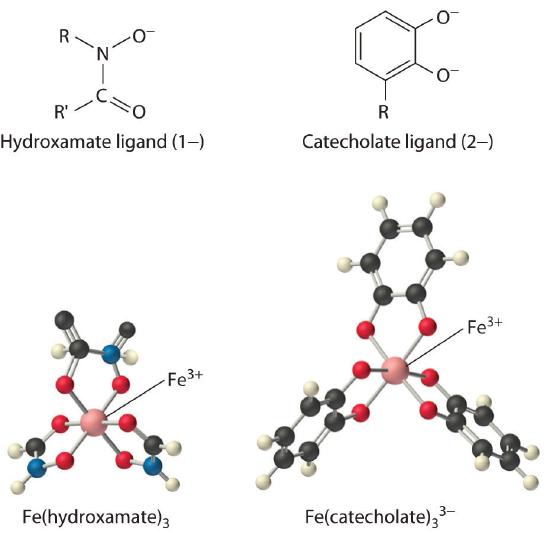
Iron complexes in biological systems. Iron(III) forms very stable octahedral complexes with hydroxamate and catecholate ligands.
The solubility of metal ions such as Fe3+, which form highly insoluble hydroxides, depends on the pH and the presence of complexing agents. In an oxygen-containing atmosphere, iron exists as Fe(III) because of the positive reduction potential of Fe3+ (Fe3+ + e− → Fe2+; E° = +0.77 V). Because ferric hydroxide [Fe(OH)3] is highly insoluble (Ksp ≈ 1 × 10−39), the equilibrium concentration of Fe3+(aq) at pH 7.0 is very low, about 10−18 M. You would have to drink 2 × 1013 L of iron-saturated water per day (roughly 5 mi3) to consume the recommended daily intake of Fe for humans, which is about 1 mg/day. Animals such as humans can overcome this problem by consuming concentrated sources of iron, such as red meat, but microorganisms cannot.
Consequently, most microorganisms synthesize and secrete organic molecules called siderophores to increase the total concentration of available iron in the surrounding medium. Siderophores are generally cyclic compounds that use bidentate ligands, such as the hydroxamate and catecholate groups shown here, to bind Fe3+ in an octahedral arrangement. Typical siderophores are ferrichrome, a cyclic peptide produced by fungi, and enterobactin, a cyclic ester produced by bacteria (Figure \(\PageIndex{1}\)). Attaching the three iron ligands to a cyclic framework greatly increases the stability of the resulting Fe3+ complex due to the chelate effect described in Section 23.4. The formation constants for the Fe3+ complexes of ferrichrome and enterobactin are about 1032 and 1040, respectively, which are high enough to allow them to dissolve almost any Fe(III) compound.
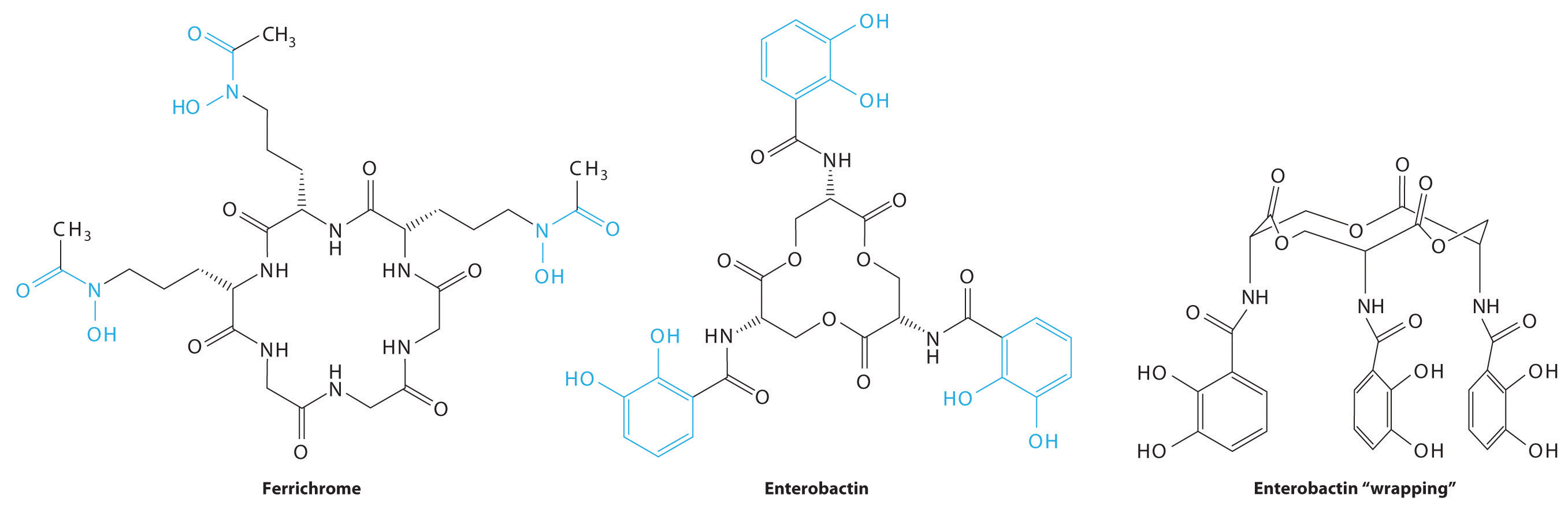
Siderophores increase the [Fe3+] in solution, providing the bacterium that synthesized them (as well as any competitors) with a supply of iron. In addition, siderophores neutralize the positive charge on the metal ion and provide a hydrophobic “wrapping” that enables the Fe3+–siderophore complex to be recognized by a specific protein that transports it into the interior of a cell. Once it is inside a cell, the iron is reduced to Fe2+, which has a much lower affinity for the siderophore and spontaneously dissociates.
In contrast, multicellular organisms can increase the concentration of iron in their diet by lowering the pH in the gastrointestinal tract. At pH 1.0 (the approximate pH of the stomach), most Fe(III) salts dissolve to form Fe3+(aq), which is absorbed by specific proteins in the intestinal wall. A protein called transferrin forms a complex with iron(III), allowing it to be transported to other cells. Proteins that bind tightly to Fe(III) can also be used as antibacterial agents because iron is absolutely essential for bacterial growth. For example, milk, tears, and egg white all contain proteins similar to transferrin, and their high affinity for Fe3+ allows them to sequester iron, thereby preventing bacteria from growing in these nutrient-rich media.
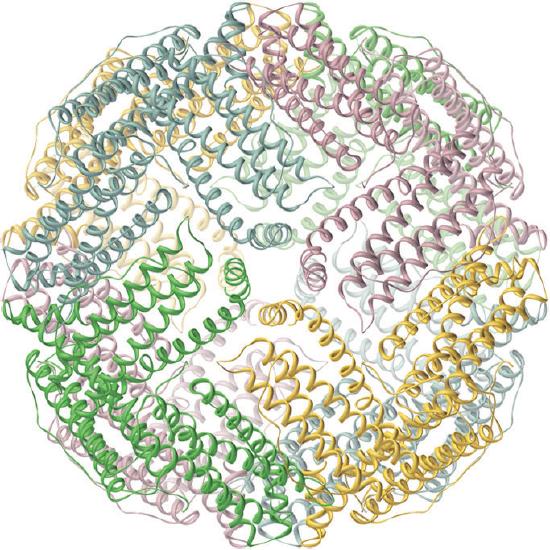
Iron is released from transferrin by reduction to Fe2+, and then it is either used immediately (e.g., for the synthesis of hemoglobin) or stored in a very large protein called ferritin for future use (Figure \(\PageIndex{2}\)). Ferritin uses oxygen to oxidize Fe2+ to Fe3+, which at neutral pH precipitates in the central cavity of the protein as a polymeric mixture of Fe(OH)3 and FePO4. Because a fully loaded ferritin molecule can contain as many as 4500 Fe atoms, which corresponds to about 25% Fe by mass, ferritin is an effective way to store iron in a highly concentrated form. When iron is needed by a cell, the Fe3+ is reduced to the much more soluble Fe2+ by a reductant such as ascorbic acid (vitamin C). The structure of ferritin contains channels at the junctions of the subunits, which provide pathways for iron to enter and leave the interior of a molecule.
Metalloproteins and Metalloenzymes
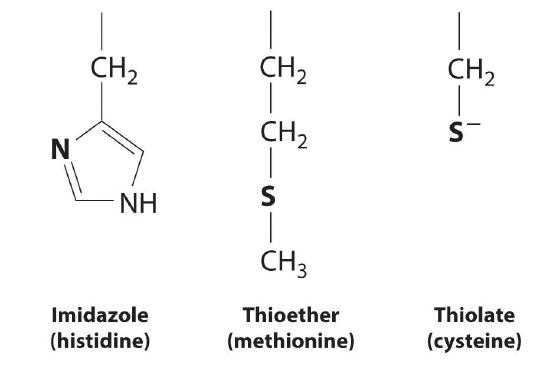
A protein that contains one or more metal ions tightly bound to amino acid side chains is called a metalloprotein; some of the most common ligands provided by amino acids are shown here. A metalloprotein that catalyzes a chemical reaction is a metalloenzyme. Thus all metalloenzymes are metalloproteins, but the converse is not true. Recent estimates suggest that more than 40% of all known enzymes require at least one metal ion for activity, including almost all the enzymes responsible for the synthesis, duplication, and repair of DNA (deoxyribonucleic acid) and RNA (ribonucleic acid).
Electron-Transfer Proteins
Proteins whose function is to transfer electrons from one place to another are called electron-transfer proteins. Because they do not catalyze a chemical reaction, electron-transfer proteins are not enzymes; they are biochemical reductants or oxidants consumed in an enzymatic reaction. The general reaction for an electron-transfer protein is as follows:
Because many transition metals can exist in more than one oxidation state, electron-transfer proteins usually contain one or more metal ions that can undergo a redox reaction. Incorporating a metal ion into a protein has three important biological consequences:
- The protein environment can adjust the redox potential (E0′), of the metal ion over a rather large potential range, whereas the redox potential of the simple hydrated metal ion [Mn+(aq)], is essentially fixed.
- The protein can adjust the structure of the metal complex to ensure that electron transfer is rapid.
- The protein environment provides specificity, ensuring that the electron is transferred to only the desired site.
Three important classes of metalloproteins transfer electrons: blue copper proteins, cytochromes, and iron–sulfur proteins, which generally transfer electrons at high (> 0.20 V), intermediate (±0 V), and low (−0.20 to −0.50 V) potentials, respectively (Table 23.12). Although these electron-transfer proteins contain different metals with different structures, they are all designed to ensure rapid electron transfer to and from the metal. Thus when the protein collides with its physiological oxidant or reductant, electron transfer can occur before the two proteins diffuse apart. For electron transfer to be rapid, the metal sites in the oxidized and reduced forms of the protein must have similar structures.
| Protein | Metal Center | M/e− Transferred | Reduction Potential (V) |
|---|---|---|---|
| * A sulfur bound to an organic group is represented as SR. | |||
| † See Figure \(\PageIndex{2b}\): for the structure of imidazole (Im). | |||
| iron–sulfur proteins* | [Fe(SR)4]2− | 1 Fe | −0.1 to +0.1 |
| [(RS)2FeS2Fe(SR)2]2− | 2 Fe | −0.2 to −0.4 | |
| [Fe3S4(SR)3]3− | 3 Fe | −0.1 to −0.2 | |
| [Fe4S4(SR)4]2− | 4 Fe | −0.3 to −0.5 | |
| cytochromes | Fe-heme (low spin) | 1 Fe | ~0 |
| blue copper proteins† | [Cu(Im)2(SR)(SR2)]− | 1 Cu | ≥ +0.20 |
Blue Copper Proteins
Blue copper proteins were first isolated from bacteria in the 1950s and from plant tissues in the early 1960s. The intense blue color of these proteins is due to a strong absorption band at a wavelength of about 600 nm. Although simple Cu2+ complexes, such as [Cu(H2O)6]2+ and [Cu(NH3)4]2+, are also blue due to an absorption band at 600 nm, the intensity of the absorption band is about 100 times less than that of a blue copper protein. Moreover, the reduction potential for the Cu2+/Cu+ couple in a blue copper protein is usually +0.3 to +0.5 V, considerably more positive than that of the aqueous Cu2+/Cu+ couple (+0.15 V).
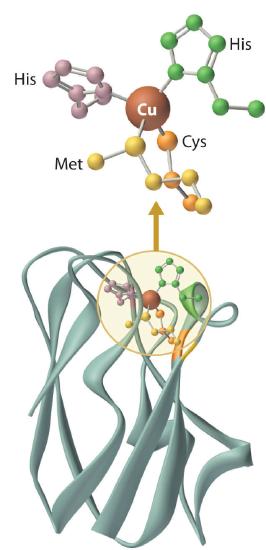
The copper center in blue copper proteins has a distorted tetrahedral structure, in which the copper is bound to four amino acid side chains (Figure \(\PageIndex{3}\)). Although the most common structures for four-coordinate Cu2+ and Cu+ complexes are square planar and tetrahedral, respectively, the structures of the oxidized (Cu2+) and reduced (Cu+) forms of the protein are essentially identical. Thus the protein forces the Cu2+ ion to adopt a higher-energy structure that is more suitable for Cu+, which makes the Cu2+ form easier to reduce and raises its reduction potential.
Moreover, by forcing the oxidized and reduced forms of the metal complex to have essentially the same structure, the protein ensures that electron transfer to and from the copper site is rapid because only minimal structural reorganization of the metal center is required. Kinetics studies on simple metal complexes have shown that electron-transfer reactions tend to be slow when the structures of the oxidized and reduced forms of a metal complex are very different, and fast when they are similar. You will see that other metal centers used for biological electron-transfer reactions are also set up for minimal structural reorganization after electron transfer, which ensures the rapid transfer of electrons.
Cytochromes
The cytochromes (from the Greek cytos, meaning “cell”, and chroma, meaning “color”) were first identified in the 1920s by spectroscopic studies of cell extracts. Based on the wavelength of the maximum absorption in the visible spectrum, they were classified as cytochromes a (with the longest wavelength), cytochromes b (intermediate wavelength), and cytochromes c (shortest wavelength). It quickly became apparent that there was a correlation between their spectroscopic properties and other physical properties. For examples, cytochromes c are generally small, soluble proteins with a reduction potential of about +0.25 V, whereas cytochromes b are larger, less-soluble proteins with reduction potentials of about 0 V.
All cytochromes contain iron, and the iron atom in all cytochromes is coordinated by a planar array of four nitrogen atoms provided by a cyclic tetradentate ligand called a porphyrin. The iron–porphyrin unit is called a heme group. The structures of a typical porphyrin (protoporphyrin IX) and its iron complex (protoheme) are shown here. In addition to the four nitrogen atoms of the porphyrin, the iron in a cytochrome is usually bonded to two additional ligands provided by the protein, as shown in Figure \(\PageIndex{4}\).
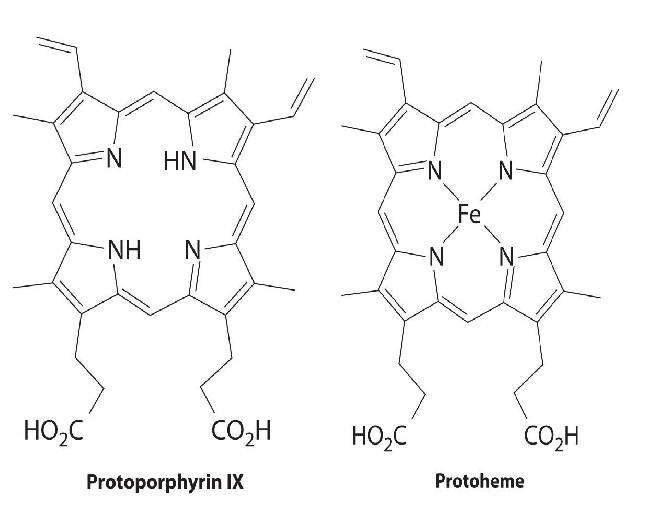
A cytochrome. Shown here is protoporphyrin IX and its iron complex, protoheme.
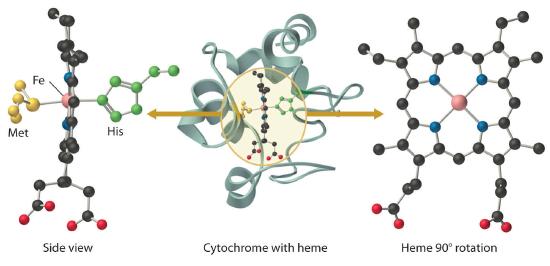
In contrast to the blue copper proteins, two electron configurations are possible for both the oxidized and reduced forms of a cytochrome, and this has significant structural consequences. Thus Fe2+ is d6 and can be either high spin (with four unpaired electrons) or low spin (with no unpaired electrons). Similarly, Fe3+ is d5 and can also be high spin (with five unpaired electrons) or low spin (with one unpaired electron). In low-spin heme complexes, both the Fe2+ and the Fe3+ ions are small enough to fit into the “hole” in the center of the porphyrin; hence the iron atom lies almost exactly in the plane of the four porphyrin nitrogen atoms in both cases. Because cytochromes b and c are low spin in both their oxidized and reduced forms, the structures of the oxidized and reduced cytochromes are essentially identical. Hence minimal structural changes occur after oxidation or reduction, which makes electron transfer to or from the heme very rapid.
Electron transfer reactions occur most rapidly when minimal structural changes occur during oxidation or reduction.
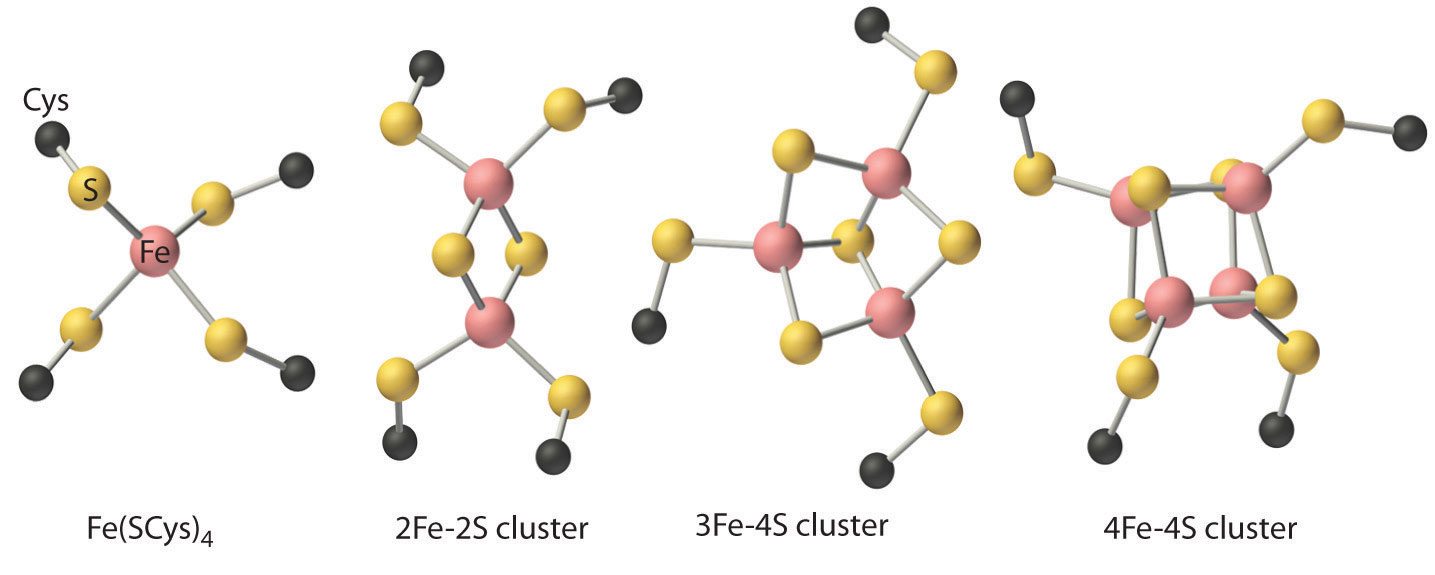
Iron–Sulfur Proteins
Although all known bacteria, plants, and animals use iron–sulfur proteins to transfer electrons, the existence of these proteins was not recognized until the late 1950s. Iron–sulfur proteins transfer electrons over a wide range of reduction potentials, and their iron content can range from 1 to more than 12 Fe atoms per protein molecule. In addition, most iron–sulfur proteins contain stoichiometric amounts of sulfide (S2−).
These properties are due to the presence of four different kinds of iron–sulfur units, which contain one, two, three, or four iron atoms per Fe–S complex (Figure \(\PageIndex{5}\)). In all cases, the Fe2+ and Fe3+ ions are coordinated to four sulfur ligands in a tetrahedral environment. Due to tetrahedral coordination by weak-field sulfur ligands, the iron is high spin in both the Fe3+ and Fe2+ oxidation states, which results in similar structures for the oxidized and reduced forms of the Fe–S complexes. Consequently, only small structural changes occur after oxidation or reduction of the Fe–S center, which results in rapid electron transfer.
Reactions of Small Molecules
Although small molecules, such as O2, N2, and H2, do not react with organic compounds under ambient conditions, they do react with many transition-metal complexes. Consequently, virtually all organisms use metalloproteins to bind, transport, and catalyze the reactions of these molecules. Probably the best-known example is hemoglobin, which is used to transport O2 in many multicellular organisms.
Under ambient conditions, small molecules, such as O2, N2, and H2, react with transition-metal complexes but not with organic compounds.
Oxygen Transport
Many microorganisms and most animals obtain energy by respiration, the oxidation of organic or inorganic molecules by O2. At 25°C, however, the concentration of dissolved oxygen in water in contact with air is only about 0.25 mM. Because of their high surface area-to-volume ratio, aerobic microorganisms can obtain enough oxygen for respiration by passive diffusion of O2 through the cell membrane. As the size of an organism increases, however, its volume increases much more rapidly than its surface area, and the need for oxygen depends on its volume. Consequently, as a multicellular organism grows larger, its need for O2 rapidly outstrips the supply available through diffusion. Unless a transport system is available to provide an adequate supply of oxygen for the interior cells, organisms that contain more than a few cells cannot exist. In addition, O2 is such a powerful oxidant that the oxidation reactions used to obtain metabolic energy must be carefully controlled to avoid releasing so much heat that the water in the cell boils. Consequently, in higher-level organisms, the respiratory apparatus is located in internal compartments called mitochondria, which are the power plants of a cell. Oxygen must therefore be transported not only to a cell but also to the proper compartment within a cell.
Three different chemical solutions to the problem of oxygen transport have developed independently in the course of evolution, as indicated in Table \(\PageIndex{2}\). Mammals, birds, reptiles, fish, and some insects use a heme protein called hemoglobin to transport oxygen from the lungs to the cells, and they use a related protein called myoglobin to temporarily store oxygen in the tissues. Several classes of invertebrates, including marine worms, use an iron-containing protein called hemerythrin to transport oxygen, whereas other classes of invertebrates (arthropods and mollusks) use a copper-containing protein called hemocyanin. Despite the presence of the hem- prefix, hemerythrin and hemocyanin do not contain a metal–porphyrin complex.
| Protein | Source | M per Subunit | M per O2 Bound | Color (deoxy form) | Color (oxy form) |
|---|---|---|---|---|---|
| hemoglobin | mammals, birds, fish, reptiles, some insects | 1 Fe | 1 Fe | red-purple | red |
| hemerythrin | marine worms | 2 Fe | 2 Fe | colorless | red |
| hemocyanin | mollusks, crustaceans, spiders | 2 Cu | 2 Cu | colorless | blue |
Myoglobin and Hemoglobin
Myoglobin is a relatively small protein that contains 150 amino acids. The functional unit of myoglobin is an iron–porphyrin complex that is embedded in the protein (Figure 26.8.1). In myoglobin, the heme iron is five-coordinate, with only a single histidine imidazole ligand from the protein (called the proximal histidine because it is near the iron) in addition to the four nitrogen atoms of the porphyrin. A second histidine imidazole (the distal histidine because it is more distant from the iron) is located on the other side of the heme group, too far from the iron to be bonded to it. Consequently, the iron atom has a vacant coordination site, which is where O2 binds.
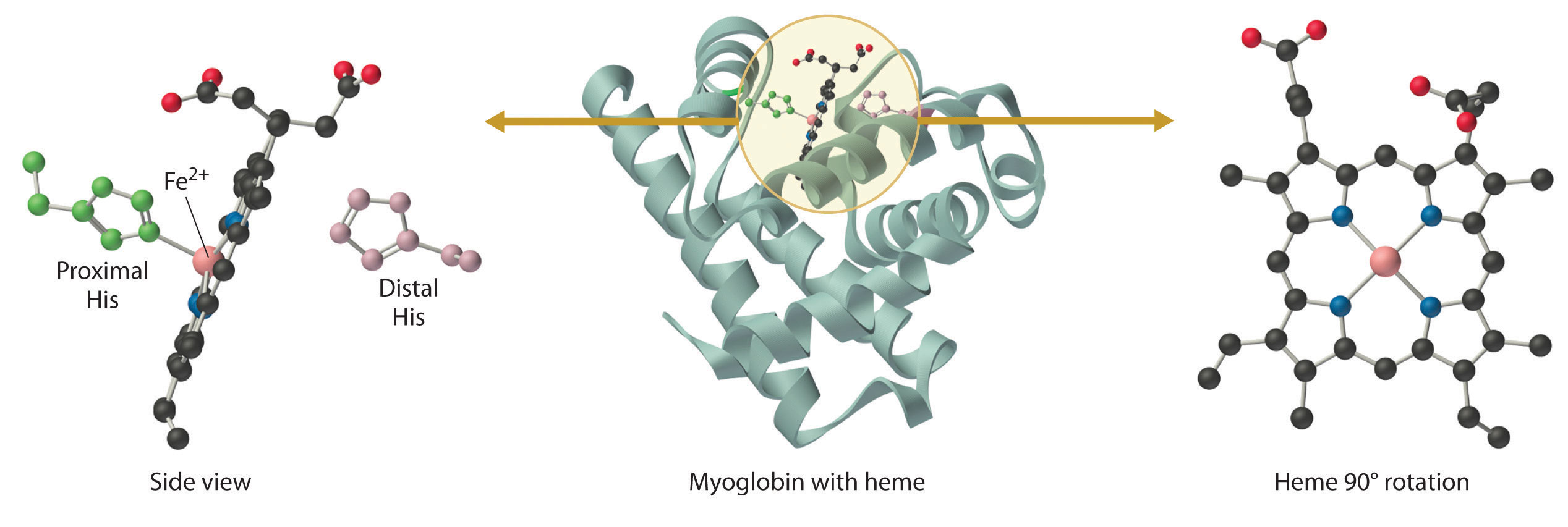
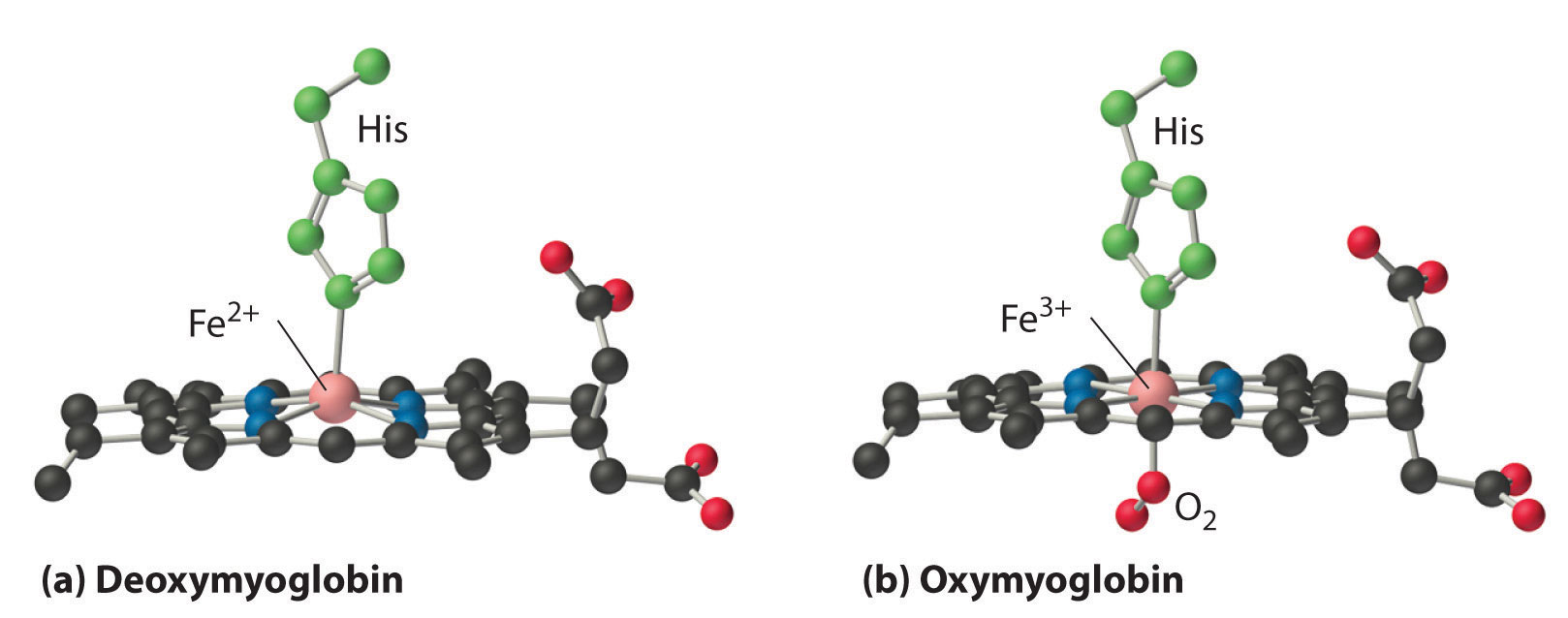
In the ferrous form (deoxymyoglobin), the iron is five-coordinate and high spin. Because high-spin Fe2+ is too large to fit into the “hole” in the center of the porphyrin, it is about 60 pm above the plane of the porphyrin. When O2 binds to deoxymyoglobin to form oxymyoglobin, the iron is converted from five-coordinate (high spin) to six-coordinate (low spin; Figure 26.8.2). Because low-spin Fe2+ and Fe3+ are smaller than high-spin Fe2+, the iron atom moves into the plane of the porphyrin ring to form an octahedral complex. The O2 pressure at which half of the molecules in a solution of myoglobin are bound to O2 (P1/2) is about 1 mm Hg (1.3 × 10−3 atm).
A vacant coordination site at a metal center in a protein usually indicates that a small molecule will bind to the metal ion, whereas a coordinatively saturated metal center is usually involved in electron transfer.
Hemoglobin consists of two subunits of 141 amino acids and two subunits of 146 amino acids, both similar to myoglobin; it is called a tetramer because of its four subunits. Because hemoglobin has very different O2-binding properties, however, it is not simply a “super myoglobin” that can carry four O2 molecules simultaneously (one per heme group). The shape of the O2-binding curve of myoglobin (Mb; Figure \(\PageIndex{7}\)) can be described mathematically by the following equilibrium:
\[MbO_2 \rightleftharpoons Mb + O_ 2 \label{26.8.1a} \]
\[K_{diss}=\dfrac{[Mb][O_2]}{[MbO_2]} \label{26.8.1b} \]
In contrast, the O2-binding curve of hemoglobin is S shaped (Figure \(\PageIndex{8}\)). As shown in the curves, at low oxygen pressures, the affinity of deoxyhemoglobin for O2 is substantially lower than that of myoglobin, whereas at high O2 pressures the two proteins have comparable O2 affinities. The physiological consequences of the unusual S-shaped O2-binding curve of hemoglobin are enormous. In the lungs, where O2 pressure is highest, the high oxygen affinity of deoxyhemoglobin allows it to be completely loaded with O2, giving four O2 molecules per hemoglobin. In the tissues, however, where the oxygen pressure is much lower, the decreased oxygen affinity of hemoglobin allows it to release O2, resulting in a net transfer of oxygen to myoglobin.
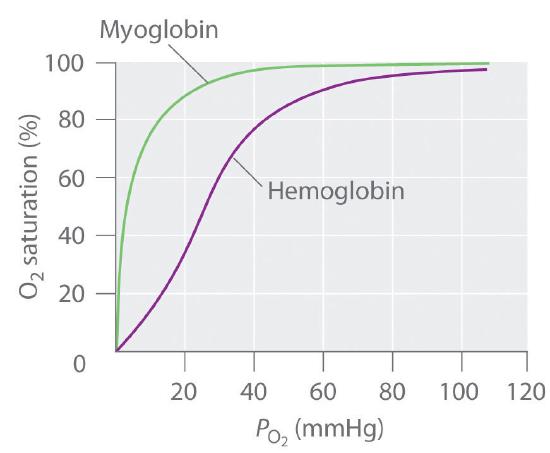
The S-shaped O2-binding curve of hemoglobin is due to a phenomenon called cooperativity, in which the affinity of one heme for O2 depends on whether the other hemes are already bound to O2. Cooperativity in hemoglobin requires an interaction between the four heme groups in the hemoglobin tetramer, even though they are more than 3000 pm apart, and depends on the change in structure of the heme group that occurs with oxygen binding. The structures of deoxyhemoglobin and oxyhemoglobin are slightly different, and as a result, deoxyhemoglobin has a much lower O2 affinity than myoglobin, whereas the O2 affinity of oxyhemoglobin is essentially identical to that of oxymyoglobin. Binding of the first two O2 molecules to deoxyhemoglobin causes the overall structure of the protein to change to that of oxyhemoglobin; consequently, the last two heme groups have a much higher affinity for O2 than the first two.
Oxygen is not unique in its ability to bind to a ferrous heme complex; small molecules such as CO and NO bind to deoxymyoglobin even more tightly than does O2. The interaction of the heme iron with oxygen and other diatomic molecules involves the transfer of electron density from the filled t2g orbitals of the low-spin d6 Fe2+ ion to the empty π* orbitals of the ligand. In the case of the Fe2+–O2 interaction, the transfer of electron density is so great that the Fe–O2 unit can be described as containing low-spin Fe3+ (d5) and O2−. We can therefore represent the binding of O2 to deoxyhemoglobin and its release as a reversible redox reaction:
\[Fe^{2+} + O_2 \rightleftharpoons \ce{Fe^{3+}–O_2^−} \label{26.8.2} \]
As shown in Figure \(\PageIndex{9}\), the Fe–O2 unit is bent, with an Fe–O–O angle of about 130°. Because the π* orbitals in CO are empty and those in NO are singly occupied, these ligands interact more strongly with Fe2+ than does O2, in which the π* orbitals of the neutral ligand are doubly occupied.
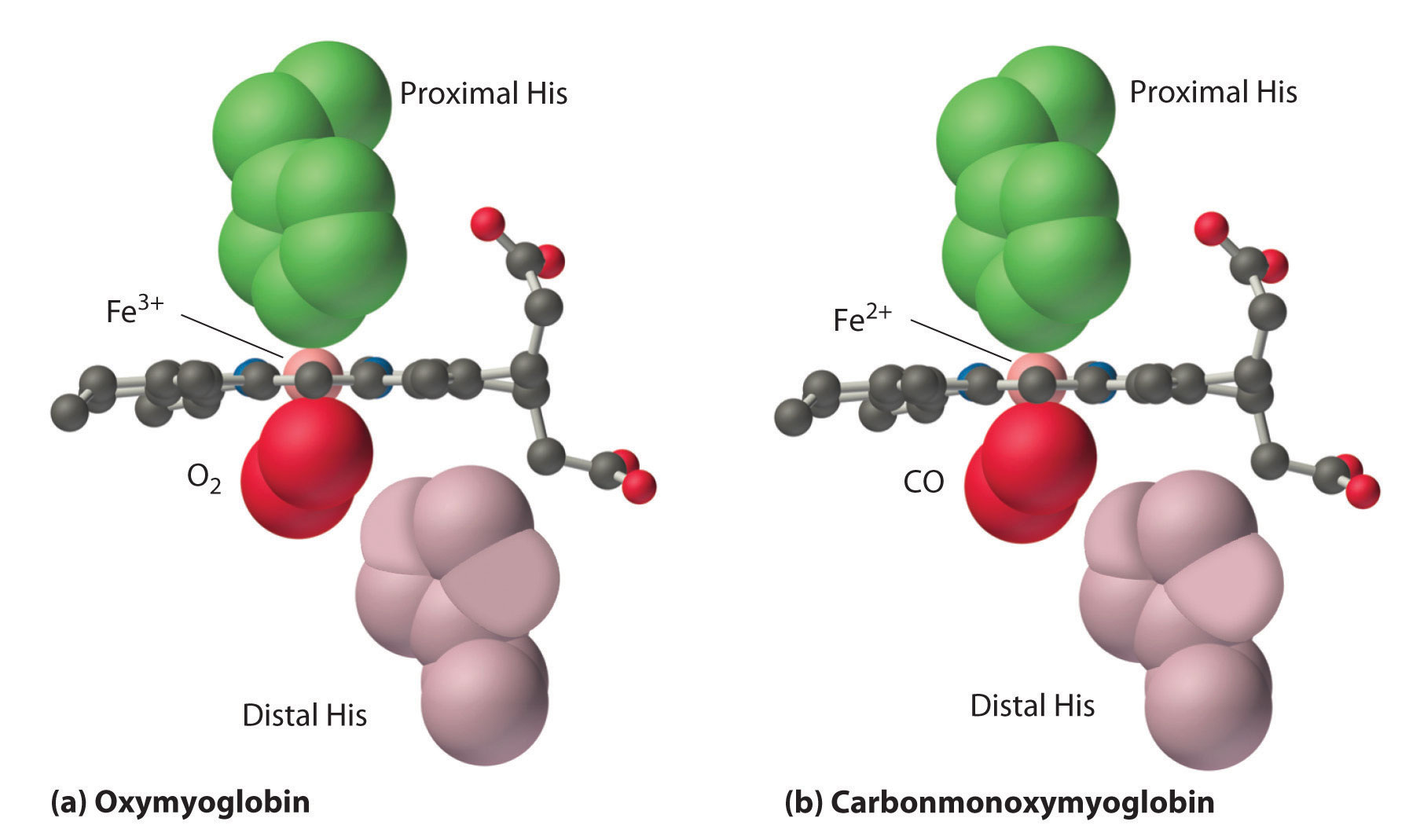
Although CO has a much greater affinity for a ferrous heme than does O2 (by a factor of about 25,000), the affinity of CO for deoxyhemoglobin is only about 200 times greater than that of O2, which suggests that something in the protein is decreasing its affinity for CO by a factor of about 100. Both CO and NO bind to ferrous hemes in a linear fashion, with an Fe–C(N)–O angle of about 180°, and the difference in the preferred geometry of O2 and CO provides a plausible explanation for the difference in affinities. As shown in Figure \(\PageIndex{9}\), the imidazole group of the distal histidine is located precisely where the oxygen atom of bound CO would be if the Fe–C–O unit were linear. Consequently, CO cannot bind to the heme in a linear fashion; instead, it is forced to bind in a bent mode that is similar to the preferred structure for the Fe–O2 unit. This results in a significant decrease in the affinity of the heme for CO, while leaving the O2 affinity unchanged, which is important because carbon monoxide is produced continuously in the body by degradation of the porphyrin ligand (even in nonsmokers). Under normal conditions, CO occupies approximately 1% of the heme sites in hemoglobin and myoglobin. If the affinity of hemoglobin and myoglobin for CO were 100 times greater (due to the absence of the distal histidine), essentially 100% of the heme sites would be occupied by CO, and no oxygen could be transported to the tissues. Severe carbon-monoxide poisoning, which is frequently fatal, has exactly the same effect. Thus the primary function of the distal histidine appears to be to decrease the CO affinity of hemoglobin and myoglobin to avoid self-poisoning by CO.
Hemerythrin
Hemerythrin is used to transport O2 in a variety of marine invertebrates. It is an octamer (eight subunits), with each subunit containing two iron atoms and binding one molecule of O2. Deoxyhemerythrin contains two Fe2+ ions per subunit and is colorless, whereas oxyhemerythrin contains two Fe3+ ions and is bright reddish violet. These invertebrates also contain a monomeric form of hemerythrin that is located in the tissues, analogous to myoglobin. The binding of oxygen to hemerythrin and its release can be described by the following reaction, where the HO2− ligand is the hydroperoxide anion derived by the deprotonation of hydrogen peroxide (H2O2):
\[\ce{2Fe^{2+} + O2 + H^{+} <=> 2Fe^{3+}–O2H} \label{23.17} \]
Thus O2 binding is accompanied by the transfer of two electrons (one from each Fe2+) and a proton to O2.
Hemocyanin
Hemocyanin is used for oxygen transport in many arthropods (spiders, crabs, lobsters, and centipedes) and in mollusks (shellfish, octopi, and squid); it is responsible for the bluish-green color of their blood. The protein is a polymer of subunits that each contain two copper atoms (rather than iron), with an aggregate molecular mass of greater than 1,000,000 amu. Deoxyhemocyanin contains two Cu+ ions per subunit and is colorless, whereas oxyhemocyanin contains two Cu2+ ions and is bright blue. As with hemerythrin, the binding and release of O2 correspond to a two-electron reaction:
\[\ce{2Cu^{+} + O2 <=> Cu^{2+}–O2^{2−}–Cu^{2+}} \label{23.18} \]
Although hemocyanin and hemerythrin perform the same basic function as hemoglobin, these proteins are not interchangeable. In fact, hemocyanin is so foreign to humans that it is one of the major factors responsible for the common allergies to shellfish.
Myoglobin, hemoglobin, hemerythrin, and hemocyanin all use a transition-metal complex to transport oxygen.
Enzymes Involved in Oxygen Activation
Many of the enzymes involved in the biological reactions of oxygen contain metal centers with structures that are similar to those used for O2 transport. Many of these enzymes also contain metal centers that are used for electron transfer, which have structures similar to those of the electron-transfer proteins discussed previously. In this section, we briefly describe two of the most important examples: dioxygenases and methane monooxygenase.
Dioxygenases are enzymes that insert both atoms of O2 into an organic molecule. In humans, dioxygenases are responsible for cross-linking collagen in connective tissue and for synthesizing complex organic molecules called prostaglandins, which trigger inflammation and immune reactions. Iron is by far the most common metal in dioxygenases; and the target of the most commonly used drug in the world, aspirin, is an iron enzyme that synthesizes a specific prostaglandin. Aspirin inhibits this enzyme by binding to the iron atom at the active site, which prevents oxygen from binding.
Methane monooxygenase catalyzes the conversion of methane to methanol. The enzyme is a monooxygenase because only one atom of O2 is inserted into an organic molecule, while the other is reduced to water:
\[\ce{CH_4 + O_2 + 2e^- + 2H^+ \rightarrow CH_3OH + H_2O} \label{23.19} \]
Because methane is the major component of natural gas, there is enormous interest in using this reaction to convert methane to a liquid fuel (methanol) that is much more convenient to ship and store. Because the C–H bond in methane is one of the strongest C–H bonds known, however, an extraordinarily powerful oxidant is needed for this reaction. The active site of methane monooxygenase contains two Fe atoms that bind O2, but the details of how the bound O2 is converted to such a potent oxidant remain unclear.
Metal Ions as Lewis Acids
Reactions catalyzed by metal ions that do not change their oxidation states during the reaction are usually group transfer reactions, in which a group such as the phosphoryl group (−PO32−) is transferred. These enzymes usually use metal ions such as Zn2+, Mg2+, and Mn2+, and they range from true metalloenzymes, in which the metal ion is tightly bound, to metal-activated enzymes, which require the addition of metal ions for activity. Because tight binding is usually the result of specific metal–ligand interactions, metalloenzymes tend to be rather specific for a particular metal ion. In contrast, the binding of metal ions to metal-activated enzymes is largely electrostatic in nature; consequently, several different metal ions with similar charges and sizes can often be used to give an active enzyme.
Metalloenzymes generally contain a specific metal ion, whereas metal-activated enzymes can use any of several metal ions of similar size and charge.
A metal ion that acts as a Lewis acid can catalyze a group transfer reaction in many different ways, but we will focus on only one of these, using a zinc enzyme as an example. Carbonic anhydrase is found in red blood cells and catalyzes the reaction of CO2 with water to give carbonic acid.
\[\ce{CO_2(g) + H_2O(l) \rightleftharpoons H^{+}(aq) + HCO^{-}3(aq)} \label{23.20} \]
Although this reaction occurs spontaneously in the absence of a catalyst, it is too slow to absorb all the CO2 generated during respiration. Without a catalyst, tissues would explode due to the buildup of excess CO2 pressure. Carbonic anhydrase contains a single Zn2+ ion per molecule, which is coordinated by three histidine imidazole ligands and a molecule of water. Because Zn2+ is a Lewis acid, the pKa of the Zn2+–OH2 unit is about 8 versus 14 for pure water. Thus at pH 7–8, a significant fraction of the enzyme molecules contain the Zn2+–OH− group, which is much more reactive than bulk water. When carbon dioxide binds in a nonpolar site next to the Zn2+–OH− unit, it reacts rapidly to give a coordinated bicarbonate ion that dissociates from the enzyme:
\[\ce{Zn^{2+}–OH^{-} + CO_2 \rightleftharpoons Zn^{2+}–OCO_2H^- \rightleftharpoons Zn^{2+} + HCO_3^{-}} \label{23.21} \]
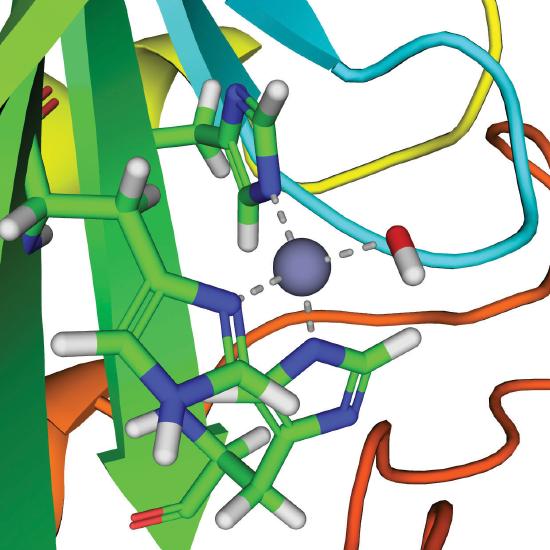
The active site of carbonic anhydrase.
Thus the function of zinc in carbonic anhydrase is to generate the hydroxide ion at pH 7.0, far less than the pH required in the absence of the metal ion.
Enzymes That Use Metals to Generate Organic Radicals
An organic radical is an organic species that contains one or more unpaired electrons. Chemists often consider organic radicals to be highly reactive species that produce undesirable reactions. For example, they have been implicated in some of the irreversible chemical changes that accompany aging. It is surprising, however, that organic radicals are also essential components of many important enzymes, almost all of which use a metal ion to generate the organic radical within the enzyme. These enzymes are involved in the synthesis of hemoglobin and DNA, among other important biological molecules, and they are the targets of pharmaceuticals for the treatment of diseases such as anemia, sickle-cell anemia, and cancer. In this section, we discuss one class of radical enzymes that use vitamin B12.
Vitamin B12 was discovered in the 1940s as the active agent in the cure of pernicious anemia, which does not respond to increased iron in the diet. Humans need only tiny amounts of vitamin B12, and the average blood concentration in a healthy adult is only about 3.5 × 10−8 M. The structure of vitamin B12, shown in Figure \(\PageIndex{10}\), is similar to that of a heme, but it contains cobalt instead of iron, and its structure is much more complex. In fact, vitamin B12 has been called the most complex nonpolymeric biological molecule known and was the first naturally occurring organometallic compound to be isolated. When vitamin B12 (the form present in vitamin tablets) is ingested, the axial cyanide ligand is replaced by a complex organic group.
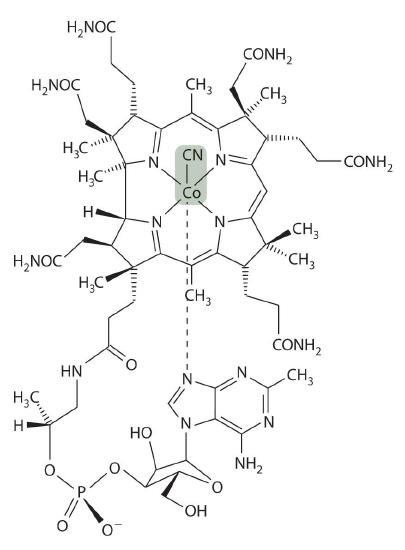
The cobalt–carbon bond in the enzyme-bound form of vitamin B12 and related compounds is unusually weak, and it is particularly susceptible to homolytic cleavage:
Homolytic cleavage of the Co3+–CH2R bond produces two species, each of which has an unpaired electron: a d7 Co2+ derivative and an organic radical, ·CH2R, which is used by vitamin B12-dependent enzymes to catalyze a wide variety of reactions. Virtually all vitamin B12-catalyzed reactions are rearrangements in which an H atom and an adjacent substituent exchange positions:
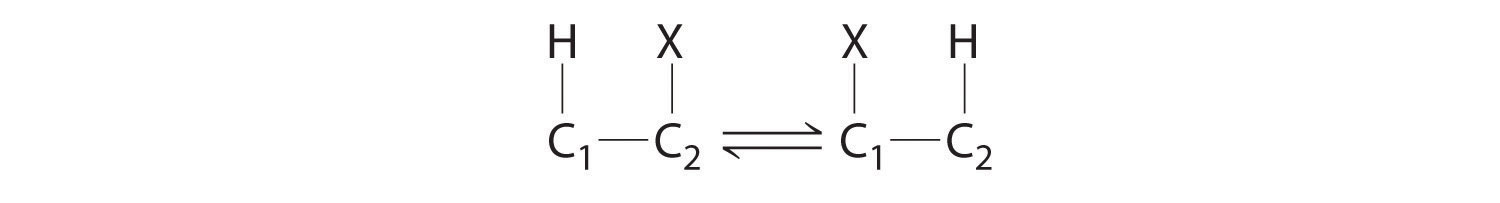
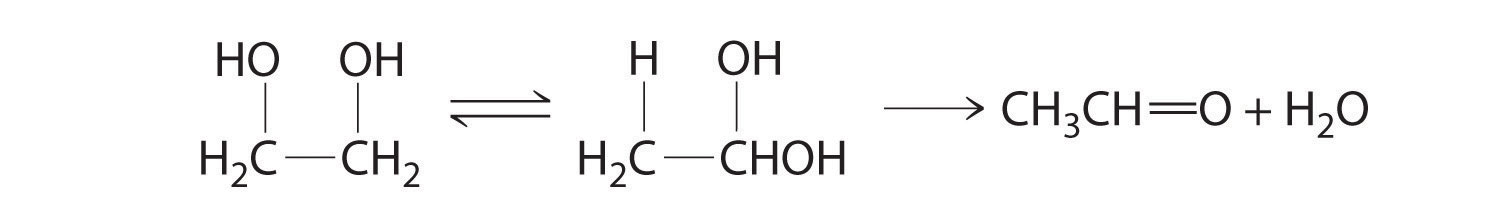
In the conversion of ethylene glycol to acetaldehyde, the initial product is the hydrated form of acetaldehyde, which rapidly loses water:
The enzyme uses the ·CH2R radical to temporarily remove a hydrogen atom from the organic substrate, which then rearranges to give a new radical. Transferring the hydrogen atom back to the rearranged radical gives the product and regenerates the ·CH2R radical.
The metal is not involved in the actual catalytic reaction; it provides the enzyme with a convenient mechanism for generating an organic radical, which does the actual work. Many examples of similar reactions are now known that use metals other than cobalt to generate an enzyme-bound organic radical.
Nearly all vitamin B12-catalyzed reactions are rearrangements that occur via a radical reaction.
Summary
Three separate steps are required for organisms to obtain essential transition metals from their environment: mobilization of the metal, transport of the metal into the cell, and transfer of the metal to where it is needed within a cell or an organism. The process of iron uptake is best understood. To overcome the insolubility of Fe(OH)3, many bacteria use organic ligands called siderophores, which have high affinity for Fe(III) and are secreted into the surrounding medium to increase the total concentration of dissolved iron. The iron–siderophore complex is absorbed by a cell, and the iron is released by reduction to Fe(II). Mammals use the low pH of the stomach to increase the concentration of dissolved iron. Iron is absorbed in the intestine, where it forms an Fe(III) complex with a protein called transferrin that is transferred to other cells for immediate use or storage in the form of ferritin.
Proteins that contain one or more tightly bound metal ions are called metalloproteins, and metalloproteins that catalyze biochemical reactions are called metalloenzymes. Proteins that transfer electrons from one place to another are called electron-transfer proteins. Most electron-transfer proteins are metalloproteins, such as iron–sulfur proteins, cytochromes, and blue copper proteins that accept and donate electrons. The oxidized and reduced centers in all electron-transfer proteins have similar structures to ensure that electron transfer to and from the metal occurs rapidly. Metalloproteins also use the ability of transition metals to bind small molecules, such as O2, N2, and H2, to transport or catalyze the reactions of these small molecules. For example, hemoglobin, hemerythrin, and hemocyanin, which contain heme iron, nonheme iron, and copper, respectively, are used by different kinds of organisms to bind and transfer O2. Other metalloenzymes use transition-metal ions as Lewis acids to catalyze group transfer reactions. Finally, some metalloenzymes use homolytic cleavage of the cobalt–carbon bond in derivatives of vitamin B12 to generate an organic radical that can abstract a hydrogen atom and thus cause molecular rearrangements to occur.
Key Takeaway
- Organisms have developed strategies to extract transition metals from the environment and use the metals in electron-transfer reactions, reactions of small molecules, Lewis-acid catalysis, and the generation of reactive organic radicals.
Conceptual Problems
- What are the advantages of having a metal ion at the active site of an enzyme?
- Why does the structure of the metal center in a metalloprotein that transfers electrons show so little change after oxidation or reduction?
Structure and Reactivity
- In enzymes, explain how metal ions are particularly suitable for generating organic radicals.
- A common method for treating carbon-monoxide poisoning is to have the patient inhale pure oxygen. Explain why this treatment is effective.