
6.7: The Helium Atom Cannot Be Solved Exactly
- Page ID
- 210829

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Adding electrons to the quantum hydrogen atom results in analytically unsolvable Schrödinger Equations (they exist, we just do not have analytical forms for them)
- A basic aspect of the corresponding multi-electron Hamiltonians is that they are NOT separable with respect to the spatial coordinate of each electron
- The solutions to multi-electron Schrödinger Equations are called multi-electron wavefunctions and they are often approximated as a product of single-electron wavefunctions (called the orbital approximation).
Multi-electron Hamiltonians
The second element in the periodic table provides our first example of a quantum-mechanical problem which cannot be solved exactly. Nevertheless, as we will show, approximation methods applied to helium can give accurate solutions in perfect agreement with experimental results. In this sense, it can be concluded that quantum mechanics is correct for atoms more complicated than hydrogen. By contrast, the Bohr theory failed miserably in attempts to apply it beyond the hydrogen atom.
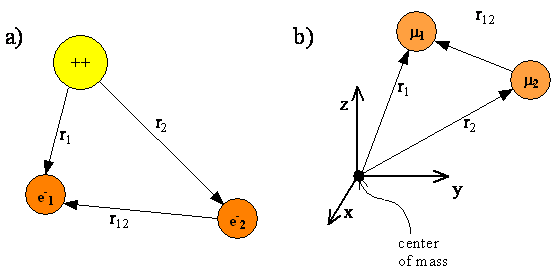
Figure \(\PageIndex{1}\) shows a schematic representation of a helium atom with two electrons whose coordinates are given by the vectors \(r_1\) and \(r_2\). The electrons are separated by a distance \(r_{12} = |r_1-r_2|\). The origin of the coordinate system is fixed at the nucleus. As with the hydrogen atom, the nuclei for multi-electron atoms are so much heavier than an electron that the nucleus is assumed to be the center of mass. Fixing the origin of the coordinate system at the nucleus allows us to exclude translational motion of the center of mass from our quantum mechanical treatment.
The Hamiltonian operator for the hydrogen atom serves as a reference point for writing the Hamiltonian operator for atoms with more than one electron. Start with the same general form we used for the hydrogen atom Hamiltonian
\[\hat {H} = \hat {T} + \hat {V} \label {6.7.1}\]
Include a kinetic energy term for each electron and a potential energy term for the attraction of each negatively charged electron for the positively charged nucleus and a potential energy term for the mutual repulsion of each pair of negatively charged electrons. The He atom Hamiltonian is
\[ \hat {H} = -\dfrac {\hbar ^2}{2m_e} (\nabla ^2_1 + \nabla ^2_2) + V_1 (r_1) + V_2 (r_2) + V_{12} (r_{12}) \label {6.7.2}\]
where
\[ V_1(r_1) = -\dfrac {2e^2}{4 \pi \epsilon _0 r_1} \label {6.7.3}\]
\[ V_2(r_2) = -\dfrac {2e^2}{4 \pi \epsilon _0 r_2} \label {6.7.4}\]
\[ V_{12}(r_{12}) = \dfrac {e^2}{4 \pi \epsilon _0 r_{12}} \label {6.7.5}\]
The two-electron Hamiltonian in Equation \(\ref{6.7.2}\) can be extended to any atom or ion by replacing the He nuclear charge of +2 with a general charge \(Z\); e.g.
\[V_1(r_1) = -\dfrac {Ze^2}{4 \pi \epsilon _0 r_1} \label {6.7.6}\]
and including terms for the additional electrons. The subsequent multi-electron atom with \(n\) electron is
\[\hat {H} = \underbrace{-\dfrac {\hbar ^2}{2m_e} \sum_i^n \nabla ^2_i}_{\text{Kinetic Energy}} + \underbrace{\sum_i^n V_i (r_i)}_{\text{Coulombic Attraction}} + \underbrace{ \sum_{i \ne j}^{n,n} V_{ij} (r_{ij})}_{\text{electron-electron Repulsion}} \label {6.7.7}\]
This multi-electron Hamiltonian is qualitatively similar to the 2-electron Hamiltonian (Equation \ref{6.7.1}) with each electron having its own kinetic energy and nuclear potential energy terms (Equations \(\ref{6.7.3}\) and \(\ref{6.7.4}\)). The other big difference between single electron systems and multi-electron systems is the presence of the \(V_{ij}(r_{ij})\) terms which contain \(1/r_{ij}\), where \(r_{ij}\) is the distance between electrons \(i\) and \(j\). These terms account for the electron-electron repulsion that we expect between like-charged particles.
For the generalized multi-electron atom Hamiltonian (Equation \(\ref{6.7.7}\)):
- Explain the origin of each of the three summations.
- What do these summations over (i.e., what is the origin of the summing index)?
- Write expressions for \(V_i(r_i)\) and \(V_{ij}(r_{ij})\).
Boron is the fifth element of the periodic table (Z=5) and is located in Group 13.
- Write the multi-electron Hamiltonian for a \(\ce{^{11}B}\) atom.
- Would it be any different for a \(\ce{^{11}B^{+}}\) ion?
- Would it be any different for a \(\ce{^{10}B}\) atom?
- Answer
-
a.
\[\hat {H}_{\ce{B}}(\vec r_1,\vec r_2,\vec r_3,\vec r_4,\vec r_5) = -\dfrac {\hbar ^2}{2m_e} \sum_i^{5} \nabla ^2_i + \sum_i^{5} \dfrac {-5e^2}{4 \pi \epsilon _0 r_i} + \sum_{i \ne j}^{5,5} \dfrac {e^2}{4 \pi \epsilon _0 r_{ij}} \nonumber\]
which expands to 20 terms
\[ \begin{align*} \hat {H}_B(\vec r_1,\vec r_2,\vec r_3,\vec r_4,\vec r_5) = &-\dfrac {\hbar ^2}{2m_e} \nabla ^2_1 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_2 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_3 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_4 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_5 \\[4pt] &- \dfrac {-5e^2}{4 \pi \epsilon _0 r_1} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_2} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_3} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_4} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_5} \\[4pt] &+ \dfrac {e^2}{4 \pi \epsilon _0 r_{12}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{13}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{14}} +\dfrac {e^2}{4 \pi \epsilon _0 r_{15}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{23}} +\dfrac {e^2}{4 \pi \epsilon _0 r_{24}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{25}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{34}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{35}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{45}} \end{align*}\]
b. Yes, \(\ce{^{11}B^{+}}\) has one less electron than \(\ce{^{11}B}\). Its Hamiltonian is
\[\hat {H}_{\ce{B^+}}(\vec r_1,\vec r_2,\vec r_3,\vec r_4 ) = -\dfrac {\hbar ^2}{2m_e} \sum_i^{4} \nabla ^2_i + \sum_i^{4} \dfrac {-5e^2}{4 \pi \epsilon _0 r_i} + \sum_{i \ne j}^{4,4} \dfrac {e^2}{4 \pi \epsilon _0 r_{ij}} \nonumber\]
or expanded to 14 terms
\[ \begin{align*} \hat {H}_B(\vec r_1,\vec r_2,\vec r_3,\vec r_4) = &-\dfrac {\hbar ^2}{2m_e} \nabla ^2_1 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_2 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_3 -\dfrac {\hbar ^2}{2m_e} \nabla ^2_4 \\[4pt] &- \dfrac {-5e^2}{4 \pi \epsilon _0 r_1} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_2} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_3} - \dfrac {-5e^2}{4 \pi \epsilon _0 r_4} \\[4pt] &+ \dfrac {e^2}{4 \pi \epsilon _0 r_{12}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{13}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{14}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{23}} +\dfrac {e^2}{4 \pi \epsilon _0 r_{24}} + \dfrac {e^2}{4 \pi \epsilon _0 r_{34}} \end{align*}\]
c. No effect. Changing the number of neutrons in the nucleus does not affect kinetic nor potential energies of the electrons. The Hamilitonian for \(\ce{^{10}B}\) is identical to \(\ce{^{11}B}\). This is technically correct for this level of discussion, but as we we will see in later, if we expand the Hamiltonian with hyperfine structure the number of neutrons can play a role.
Multi-electron Wavefunctions and the Orbital Approximation
Given what we have learned from the previous quantum mechanical systems we’ve studied, we predict that exact solutions to the multi-electron Schrödinger equation in Equation \(\ref{6.7.7}\) would consist of a family of multi-electron wavefunctions, each with an associated energy eigenvalue. These wavefunctions and energies would describe the ground and excited states of the multi-electron atom, just as the hydrogen wavefunctions and their associated energies describe the ground and excited states of the hydrogen atom. We would predict quantum numbers to be involved, as well.
The fact that electrons interact through their Coulomb repulsion means that an exact wavefunction for a multi-electron system would be a single function that depends simultaneously upon the coordinates of all the electrons; i.e., a multi-electron wavefunction,
\[\psi (\vec r _1, \vec r_2, \vec r_3, \vec r_4, \cdots \vec r_n)\]
The modulus squared of such a wavefunction would describe the probability of finding the electrons (though not specific ones) at a designated volume (\(V\)) in the atom.
\[ p(V)= \int_{V} | \psi (\vec r _1, \vec r_2, \vec r_3, \vec r_4, \cdots \vec r_n) |^2 d\tau \]
All of the electrons are described simultaneously by a multi-electron wavefunction, so the total amount of electron density represented by the wavefunction equals the number of electrons in the atom.
\[ \int_{\text{all space}} | \psi (\vec r _1, \vec r_2, \vec r_3, \vec r_4, \cdots \vec r_i) |^2 d\tau = n\]
Unfortunately, the Coulomb repulsion terms (Equation \(\ref{6.7.5}\)) make it impossible to find an exact solution to the Schrödinger equation for many-electron atoms and molecules even for two electrons atoms. We have to rely on approximations and the orbital approximation is central to basic chemistry concepts.
The most basic approximations to the exact solutions to a multi-electron atom Hamiltonian, \(\hat{H}\), (Equation \ref{6.7.7}) involve writing a multi-electron wavefunction (\(\psi (r_1, r_2, \cdots , r_n)\)) as a simple product of single-electron wavefunctions (\( \varphi _i (r_i)\) ):
\[\psi (r_1, r_2, \cdots , r_n) \approx \varphi _1 (r_1) \varphi _2 (r_2) \cdots \varphi _n(r_n) \label {6.7.8} \]
or in Dirac notation
\[|\psi (r_1, r_2, \cdots , r_n) \rangle \approx | \varphi _1 (r_1) \rangle |\varphi _2 (r_2) \rangle \cdots |\varphi _n(r_n) \rangle\label {6.7.9} \]
The energy of the atom in the state associated with a specific multi-electron wavefunction (\(E\)) is obtained from the multi-electron Schrödinger Equation
\[ \hat{H} \psi (r_1, r_2, \cdots , r_n) = E \psi (r_1, r_2, \cdots , r_n) \]
Within the approximation in Equation \ref{6.7.8}, \(E\) can be expressed sum of the energies of the one-electron components (\(\epsilon_i\)).
\[ E \approx \sum_i \epsilon_i \]
This is called the orbital approximation.
By writing the multi-electron wavefunction as a product of single-electron functions (Equations \ref{6.7.8} or \ref{6.7.9}), we conceptually transform a multi-electron atom into a collection of individual electrons located in individual orbitals whose spatial characteristics and energies can be separately identified. For atoms, these single-electron wavefunctions are called atomic orbitals and resemble the wavefunctions for hydrogen-like atoms. For molecules, as we will see in the following chapters, these are called molecular orbitals. While a great deal can be learned from such an analysis, it is important to keep in mind that such a discrete, compartmentalized picture of the electrons is an approximation, albeit a powerful one.
Contributors
David M. Hanson, Erica Harvey, Robert Sweeney, Theresa Julia Zielinski ("Quantum States of Atoms and Molecules")