
22.7: Reactions of Arylamines
- Page ID
- 183156
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Overreaction of Aniline
Arylamines are very reactive towards electrophilic aromomatic substitution. The strongest activating and ortho/para-directing substituents are the amino (-NH2) and hydroxyl (-OH) groups. Direct nitration of phenol (hydroxybenzene) by dilute nitric acid gives modest yields of nitrated phenols and considerable oxidative decomposition to tarry materials; aniline (aminobenzene) is largely destroyed. Monobromination of both phenol and aniline is difficult to control, with di- and tri-bromo products forming readily.

Because of their high nucleophilic reactivity, aniline and phenol undergo substitution reactions with iodine, a halogen that is normally unreactive with benzene derivatives. The mixed halogen iodine chloride (ICl) provides a more electrophilic iodine moiety, and is effective in iodinating aromatic rings having less powerful activating substituents.
| C6H5–NH2 + I2 + NaHCO3 | p-I–C6H4–NH2 + NaI + CO2 + H2O |
In addition to overreactivity, we have previously seen that Friedel-Crafts reactions employing AlCl3 catalyst do not work with aniline. A salt complex forms and prevents electrophilic aromatic substitution.

Both this problem and the aniline overreactivity can be circumvented by first going through the corresponding amide.
Modifying the Influence of Strong Activating Groups
By acetylating the heteroatom substituent on aniline, its activating influence can be substantially attenuated. For example, acetylation of aniline gives acetanilide (first step in the following equation), which undergoes nitration at low temperature, yielding the para-nitro product in high yield. The modifying acetyl group can then be removed by acid-catalyzed hydrolysis (last step), to yield para-nitroaniline. Although the activating influence of the amino group has been reduced by this procedure, the acetyl derivative remains an ortho/para-directing and activating substituent.
C6H5–NH2 + (CH3CO)2O |
pyridine (a base) |
C6H5–NHCOCH3 |
HNO3 , 5 ºC |
p-O2N–C6H4–NHCOCH3 |
H3O(+) & heat |
p-O2N–C6H4–NH |
 .
.
Diazonium Ions and their Salts
A resonance description of diazonium ions shows that the positive charge is delocalized over the two nitrogen atoms. It is not possible for nucleophiles to bond to the inner nitrogen, but bonding (or coupling) of negative nucleophiles to the terminal nitrogen gives neutral azo compounds. As shown in the following equation, this coupling to the terminal nitrogen should be relatively fast and reversible. The azo products may exist as E / Z stereoisomers. In practice it is found that the E-isomer predominates at equilibrium.
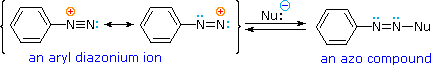
Unless these azo products are trapped or stabilized in some manner, reversal to the diazonium ion and slow nucleophilic substitution at carbon (with irreversible nitrogen loss) will be the ultimate course of reaction, as described in the previous section. For example, if phenyldiazonium bisufate is added rapidly to a cold solution of sodium hydroxide a relatively stable solution of sodium phenyldiazoate (the conjugate base of the initially formed diazoic acid) is obtained. Lowering the pH of this solution regenerates phenyldiazoic acid (pKa ca. 7), which disassociates back to the diazonium ion and eventually undergoes substitution, generating phenol.
| C6H5N2(+) HSO4(–) + NaOH (cold solution) |  |
C6H5N2–OH + NaOH (cold) | |
C6H5N2–O(–) Na(+) |
| phenyldiazonium bisulfate | phenyldiazoic acid | sodium phenyldiazoate |
Aryl diazonium salts may be reduced to the corresponding hydrazines by mild reducing agents such as sodium bisulfite, stannous chloride or zinc dust. The bisulfite reduction may proceed by an initial sulfur-nitrogen coupling, as shown in the following equation.
Ar-N2(+) X(–) |
NaHSO3 |
Ar-N=N-SO3H |
NaHSO3 |
Ar-NH-NH-SO3H |
H2O |
Ar-NH-NH2 + H2SO4 |
Diazonium Salts: The Sandmeyer Reactions
Aryl diazonium salts are important intermediates. They are prepared in cold (0 º to 10 ºC) aqueous solution, and generally react with nucleophiles with loss of nitrogen. Some of the more commonly used substitution reactions are shown in the following diagram. Since the leaving group (N2) is thermodynamically very stable, these reactions are energetically favored. Those substitution reactions that are catalyzed by cuprous salts are known as Sandmeyer reactions. Fluoride substitution occurs on treatment with BF4(–), a reaction known as the Schiemann reaction. Stable diazonium tetrafluoroborate salts may be isolated, and on heating these lose nitrogen to give an arylfluoride product. The top reaction with hypophosphorus acid, H3PO2, is noteworthy because it achieves the reductive removal of an amino (or nitro) group. Unlike the nucleophilic substitution reactions, this reduction probably proceeds by a radical mechanism.
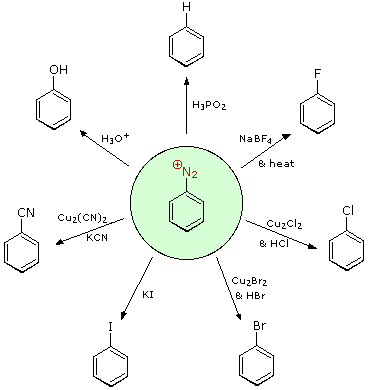
These aryl diazonium substitution reactions significantly expand the tactics available for the synthesis of polysubstituted benzene derivatives. Consider the following options:
- The usual precursor to an aryl amine is the corresponding nitro compound. A nitro substituent deactivates an aromatic ring and directs electrophilic substitution to meta locations.
- Reduction of a nitro group to an amine may be achieved in several ways. The resulting amine substituent strongly activates an aromatic ring and directs electrophilic substitution to ortho & para locations.
- The activating character of an amine substituent may be attenuated by formation of an amide derivative (reversible), or even changed to deactivating and meta-directing by formation of a quaternary-ammonium salt (irreversible).
- Conversion of an aryl amine to a diazonium ion intermediate allows it to be replaced by a variety of different groups (including hydrogen), which may in turn be used in subsequent reactions.
The following examples illustrate some combined applications of these options to specific cases. You should try to conceive a plausible reaction sequence for each. Once you have done so, you may check suggested answers below.
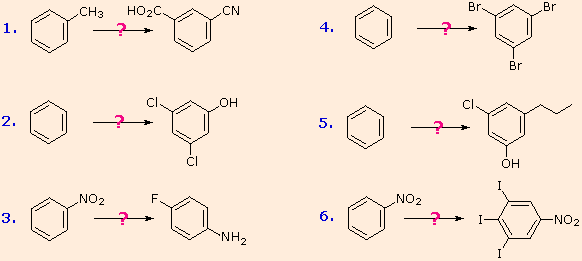
- Answer 1:
-
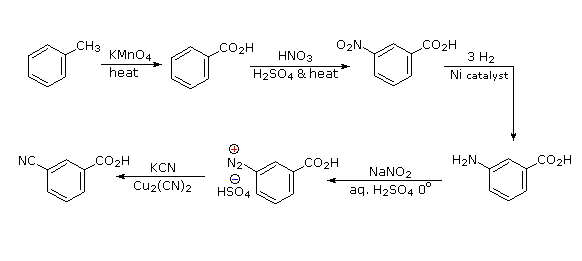
It should be clear that the methyl substituent will eventually be oxidized to a carboxylic acid function. The timing is important, since a methyl substituent is ortho/para-directing and the carboxyl substituent is meta-directing. The cyano group will be introduced by a diazonium intermediate, so a nitration followed by reduction to an amine must precede this step.
- Answer 2:
-
The hydroxyl group is a strong activating substituent and would direct aromatic ring chlorination to locations ortho & para to itself, leading to the wrong product. As an alternative, the nitro group is not only meta-directing, it can be converted to a hydroxyl group by way of a diazonium intermediate. The resulting strategy is self evident.
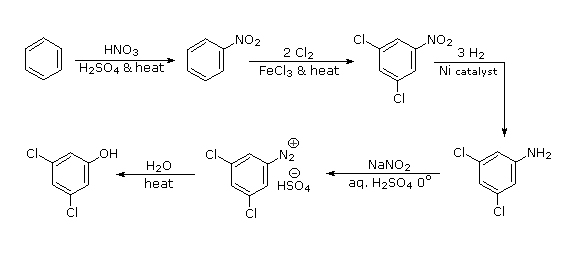
- Answer 3:
-
Selective introduction of a fluorine is best achieved by treating a diazonium intermediate with boron tetrafluoride anion. To get the necessary intermediate we need to make p-nitroaniline. Since the nitro substituent on the starting material would direct a new substituent to a meta-location, we must first reduce it to an ortho/para-directing amino group. Amino groups are powerful activating substituents, so we deactivate it by acetylation before nitration. The acetyl substituent also protects the initial amine function from reaction with nitrous acid later on. It is removed in the last step.
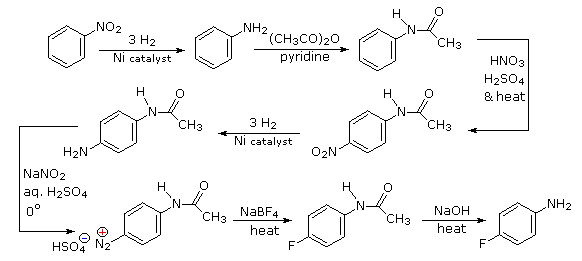
- Answer 4:
-
Polybromination of benzene would lead to ortho/para substitution. In order to achieve the mutual meta-relationship of three bromines, it is necessary to introduce a powerful ortho/para-directing prior to bromination, and then remove it following the tribromination. An amino group is ideal for this purpose. Reductive removal of the diazonium group may be accomplished in several ways (three are shown).
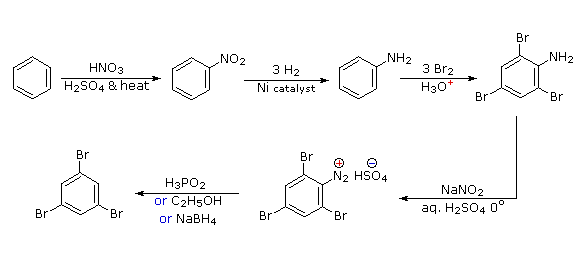
- Answer 5:
-
The propyl substituent is best introduced by Friedel-Crafts acylation followed by reduction, and this cannot be carried out in the presence of a nitro substituent. Since an acyl substituent is a meta-director, it is logical to use this property to locate the nitro and chloro groups before reducing the carbonyl moiety. The same reduction method can be used to reduce both the nitro group (to an amine) and the carbonyl group to propyl. We have already seen the use of diazonium intermediates as precursors to phenols.
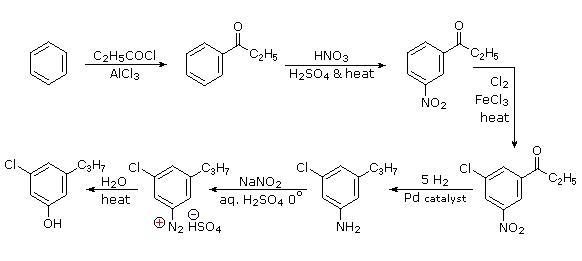
- Answer 6:
-
Aromatic iodination can only be accomplished directly on highly activated benzene compounds, such as aniline, or indirectly by way of a diazonium intermediate. Once again, a deactivated amino group is the precursor of p-nitroaniline (prb.#3). This aniline derivative requires the more electrophilic iodine chloride (ICl) for ortho-iodination because of the presence of a deactivating nitro substituent. Finally, the third iodine is introduced by the diazonium ion procedure.
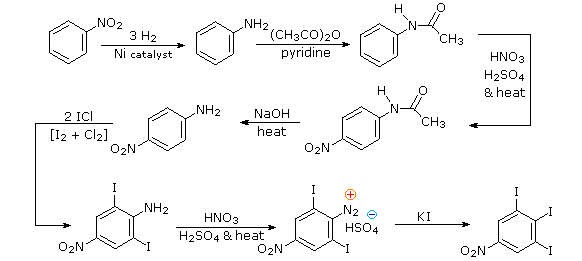
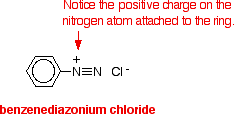
Substitution reactions of diazonium ions
Diazonium ions are present in solutions such as benzenediazonium chloride solution. They contain an -N2+ group. In the case of benzenediazonium chloride, this is attached to a benzene ring. Benzenediazonium chloride looks like this:
In this set of reactions of the diazonium ion, the -N2+ group is replaced by something else. The nitrogen is released as nitrogen gas.
Substitution by an -OH group
To get this reaction, all you need to do is warm the benzenediazonium chloride solution. The diazonium ion reacts with the water in the solution and phenol is formed - either in solution or as a black oily liquid (depending on how much is formed). Nitrogen gas is evolved.
![]()
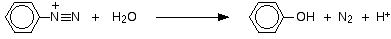
This is the same reaction that you get if you react phenylamine with nitrous acid in the warm. The diazonium ion is formed first and then immediately reacts with the water in the solution to give phenol.
Substitution by an iodine atom
This is a good example of the use of diazonium salts to substitute things into a benzene ring which are otherwise quite difficult to attach. (That's equally true of the previous reaction, by the way.) If you add potassium iodide solution to the benzenediazonium chloride solution in the cold, nitrogen gas is given off, and you get oily droplets of iodobenzene formed.
There is a simple reaction between the diazonium ions and the iodide ions from the potassium iodide solution.
![]()
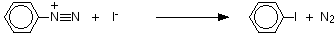
Coupling reactions of diazonium ions
In the substitution reactions above, the nitrogen in the diazonium ion is lost. In the rest of the reactions on this page, the nitrogen is retained and used to make a bridge between two benzene rings.
The most important application of diazo coupling reactions is electrophilic aromatic substitution of activated benzene derivatives by diazonium electrophiles. The products of such reactions are highly colored aromatic azo compounds that find use as synthetic dyestuffs, commonly referred to as azo dyes. Azobenzene (Y=Z=H) is light orange; however, the color of other azo compounds may range from red to deep blue depending on the nature of the aromatic rings and the substituents they carry. Azo compounds may exist as cis/trans isomer pairs, but most of the well-characterized and stable compounds are trans.
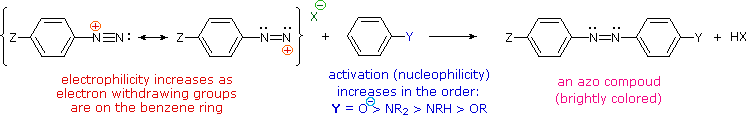
The reaction with phenol
Phenol is dissolved in sodium hydroxide solution to give a solution of sodium phenoxide.
![]()

The solution is cooled in ice, and cold benzenediazonium chloride solution is added. There is a reaction between the diazonium ion and the phenoxide ion and a yellow-orange solution or precipitate is formed. The product is one of the simplest of what are known as azo compounds, in which two benzene rings are linked by a nitrogen bridge.
![]()
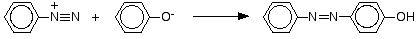
The reaction with naphthalen-2-ol
Naphthalen-2-ol is also known as 2-naphthol or beta-naphthol. It contains an -OH group attached to a naphthalene molecule rather than to a simple benzene ring. Naphthalene has two benzene rings fused together. The reaction is done under exactly the same conditions as with phenol. The naphthalen-2-ol is dissolved in sodium hydroxide solution to produce an ion just like the phenol one. This solution is cooled and mixed with the benzenediazonium chloride solution.
An intense orange-red precipitate is formed - another azo compound.
![]()
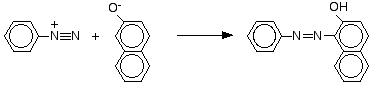
The reaction with phenylamine (aniline)
Some liquid phenylamine is added to a cold solution of benzenediazonium chloride, and the mixture is shaken vigorously. A yellow solid is produced.
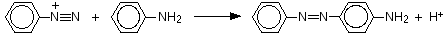
These strongly coloured azo compounds are frequently used as dyes known as azo dyes. The one made from phenylamine (aniline) is known as "aniline yellow" (amongst many other things - see note above). Azo compounds account for more than half of modern dyes.
The use of an azo dye as an indicator - methyl orange
Azo compounds contain a highly delocalized system of electrons which takes in both benzene rings and the two nitrogen atoms bridging the rings. The delocalization can also extend to things attached to the benzene rings as well. If white light falls on one of these molecules, some wavelengths are absorbed by these delocalized electrons. The colour you see is the result of the non-absorbed wavelengths. The groups which contribute to the delocalization (and so to the absorption of light) are known as a chromophore.
Modifying the groups present in the molecule can have an effect on the light absorbed, and so on the color you see. You can take advantage of this in indicators. Methyl orange is an azo dye which exists in two forms depending on the pH:
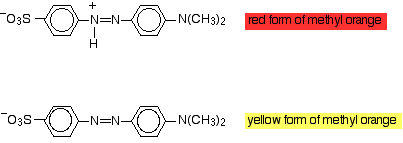
As the hydrogen ion is lost or gained there is a shift in the exact nature of the delocalisation in the molecule, and that causes a shift in the wavelength of light absorbed. Obviously that means that you see a different colour. When methyl orange is added, a hydrogen ion attaches to give the red form. Methyl orange is red in acidic solutions (in fact solutions of pH less than 3.1). If an alkali is added or hydrogen ions are removed, then the yellow form if generated. Methyl orange is yellow at pH's greater than 4.4.
In between, at some point there will be equal amounts of the red and yellow forms and so methyl orange looks orange.
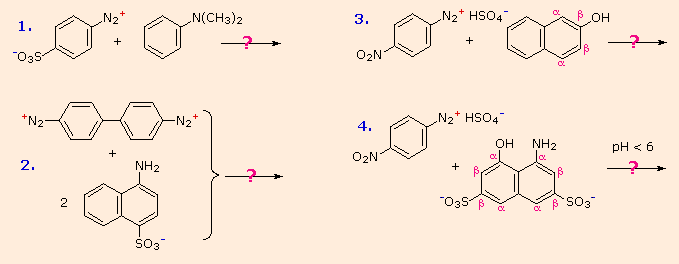
Some examples of azo coupling reactions are shown below. A few simple rules are helpful in predicting the course of such reactions:
- At acid pH (< 6) an amino group is a stronger activating substituent than a hydroxyl group (i.e. a phenol). At alkaline pH (> 7.5) phenolic functions are stronger activators, due to increased phenoxide base concentration.
- Coupling to an activated benzene ring occurs preferentially para to the activating group if that location is free. Otherwise ortho-coupling will occur.
- Naphthalene normally undergoes electrophilic substitution at an alpha-location more rapidly than at beta-sites; however, ortho-coupling is preferred. See the diagram for examples of α / β notation in naphthalenes.
You should try to conceive a plausible product structure for each of the following couplings.
Exercise
14. Propose a synthesis for the following compound via benzene and any amine you may require.
15. Proposes synthesis for each of the following compounds via benzene.
(a) N,N-Diethylaniline
(b) p-Bromoaniline
(c) m-Bromoaniline
(d) 2,4-Diethylaniline
16. Propose a synthesis for each of the following molecules from benzene via the diazonium ion.
(a) p-Chlorobenzoic acid
(b) m-Chlorobenzoic acid
(c) m-Dichlorobenzene
(d) p-Ethylbenzoic acid
(e) 1,2,4-Trichlorobenzene
Classify the following alcohols as primary, secondary, or tertiary.
- Answer
-
14.
15.
(a) 1. HNO3, H2SO4; 2. Zn(Hg), HCl; 3. EtBr
(b) 1. HNO3, H2SO4; 2. Zn(Hg), HCl; 3. (CH3CO)O2; 4. Br2, FeBr3; 5. H2O, NaOH
(c) 1. HNO3, H2SO4; 2. Br2, FeBr3; 3. Zn(Hg), HCl
(d) 1. HNO3, H2SO4; 2. Zn(Hg), HCl; 3. (CH3CO)O2; 4. EtCl, AlCl3; 5. H2O, NaOH
16.
(a) 1. CH3CH2Cl, AlCl3; 2. HNO3, H2SO4; 3. SnCl2; 4. NaNO2, H2SO4; 5. CuBr; 6. KMnO4, H2O
(b) 1. HNO3, H2SO4; 2. Cl2, FeCl3; 3. SnCl2, H3O+; 4. NaNO2, H2SO4; 5. CuCN; 6. H3O+
(c) 1. HNO3, H2SO4; 2. Cl2, FeCl3; 3. SnCl2; 4. NaNO2, H2SO4; 5. CuCl
(d) 1. CH3CH2Cl, AlCl3: 2. HNO3, H2SO4; 3. SnCl2; 4. NaNO2, H2SO4; 5. CuCN; 6. H3O+
(e) 1. HNO3, H2SO4; 2. H2/PtO2; 3. (CH3CO)2O; 4. 2 Cl2; 5. H2O, NaOH; 6. NaNO2, H2SO4; 7. CuCl
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Jim Clark (Chemguide.co.uk)