
11.5: Acidity of Alcohols and Phenols
- Page ID
- 182994
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Overview - Aqueous vs Organic Solvents
In aqueous solutions, phenols are weakly acidic and lower the pH of a solution. Sodium hydroxide can be used to fully deprotonate a phenol. Water soluble alcohols do not change the pH of the solution and are considered neutral. Aqueous solutions of sodium hydroxide can NOT deprotonate alcohols to a high enough concentration to be synthetically useful.
In solutions of organic solvents, more extreme reaction conditions can be created. Sodium metal can be added to an alcohol in an organic solvent system to fully deprotonate the alcohol to form alkoxide ions.
Acidity of Alcohols
Several important chemical reactions of alcohols involving the O-H bond or oxygen-hydrogen bond only and leave the carbon-oxygen bond intact. An important example is salt formation with acids and bases. Alcohols, like water, are both weak bases and weak acids. The acid ionization constant (Ka) of ethanol is about 10~18, slightly less than that of water. Ethanol can be converted to its conjugate base by the conjugate base of a weaker acid such as ammonia {Ka — 10~35), or hydrogen (Ka ~ 10-38). It is convenient to employ sodium metal or sodium hydride, which react vigorously but controllably with alcohols:
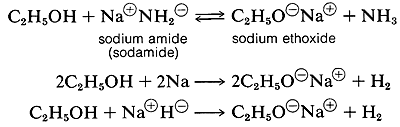
The order of acidity of various liquid alcohols generally is water > primary > secondary > tertiary ROH. By this we mean that the equilibrium position for the proton-transfer reaction lies more on the side of ROH as R is changed from primary to secondary to tertiary; therefore, tert-butyl alcohol is considered less acidic than ethanol:
\[ ROH + OH^- \rightleftharpoons RO^- + HOH\]
However, in the gas phase the order of acidity is reversed, and the equilibrium position for lies increasingly on the side of the alkoxide as R is changed from primary to secondary to tertiary, tert-butyl alcohol is therefore more acidic than ethanol in the gas phase. This seeming contradiction appears more reasonable when one considers what effect solvation (or the lack of it) has on equilibria. In solution, the larger alkoxide ions, probably are less well solvated than the smaller ions, because fewer solvent molecules can be accommodated around the negatively charged oxygen in the larger ions:
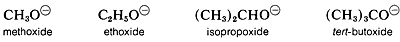
Acidity of alcohols therefore decreases as the size of the conjugate base increases. However, “naked” gaseous ions are more stable the larger the associated R groups, probably because the larger R groups can stabilize the charge on the oxygen atom better than the smaller R groups. They do this by polarization of their bonding electrons, and the bigger the group, the more polarizable it is.
Basicity of Alcohols
Alcohols are bases similar in strength to water and accept protons from strong acids. An example is the reaction of methanol with hydrogen bromide to give methyloxonium bromide, which is analogous to the formation of hydroxonium bromide with hydrogen bromide and water:

Acidity of Phenol
Compounds like alcohols and phenol which contain an -OH group attached to a hydrocarbon are very weak acids. Alcohols are so weakly acidic that, for normal lab purposes, their acidity can be virtually ignored. However, phenol is sufficiently acidic for it to have recognizably acidic properties - even if it is still a very weak acid. A hydrogen ion can break away from the -OH group and transfer to a base. For example, in solution in water:

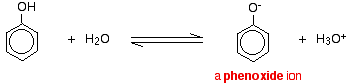
Phenol is a very weak acid and the position of equilibrium lies well to the left. Phenol can lose a hydrogen ion because the phenoxide ion formed is stabilised to some extent. The negative charge on the oxygen atom is delocalised around the ring. The more stable the ion is, the more likely it is to form. One of the lone pairs on the oxygen atom overlaps with the delocalised electrons on the benzene ring.
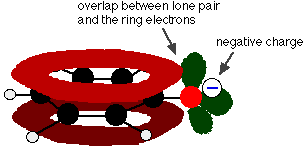
This overlap leads to a delocalization which extends from the ring out over the oxygen atom. As a result, the negative charge is no longer entirely localized on the oxygen, but is spread out around the whole ion.
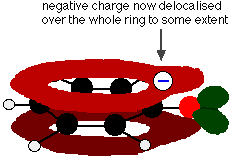
Spreading the charge around makes the ion more stable than it would be if all the charge remained on the oxygen. However, oxygen is the most electronegative element in the ion and the delocalized electrons will be drawn towards it. That means that there will still be a lot of charge around the oxygen which will tend to attract the hydrogen ion back again. That is why phenol is only a very weak acid.
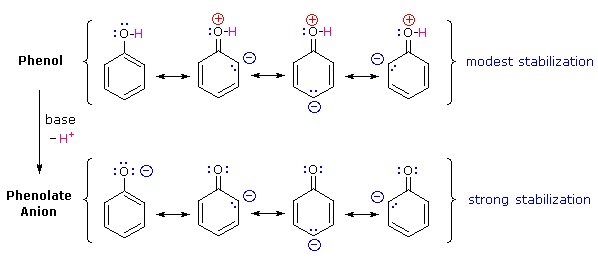
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures.
The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.

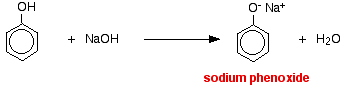
In this reaction, the hydrogen ion has been removed by the strongly basic hydroxide ion in the sodium hydroxide solution.
Acids react with the more reactive metals to give hydrogen gas. Phenol is no exception - the only difference is the slow reaction because phenol is such a weak acid. Phenol is warmed in a dry tube until it is molten, and a small piece of sodium added. There is some fizzing as hydrogen gas is given off. The mixture left in the tube will contain sodium phenoxide.

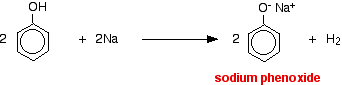
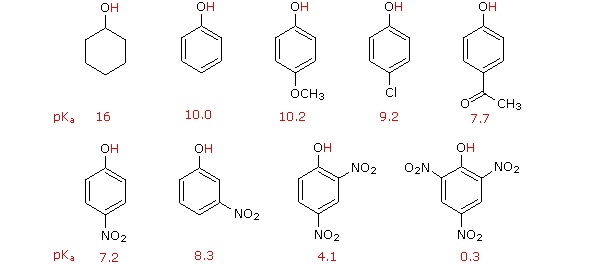
Acidity of Substituted Phenols
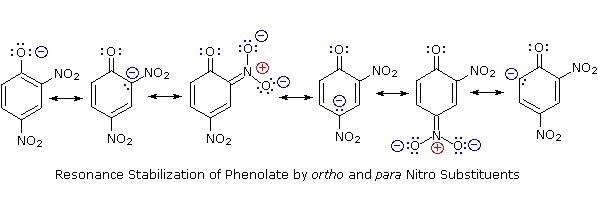
Substitution of the hydroxyl hydrogen atom is even more facile with phenols, which are roughly a million times more acidic than equivalent alcohols. This phenolic acidity is further enhanced by electron-withdrawing substituents ortho and para to the hydroxyl group, as displayed in the following diagram. The alcohol cyclohexanol is shown for reference at the top left. It is noteworthy that the influence of a nitro substituent is over ten times stronger in the para-location than it is meta, despite the fact that the latter position is closer to the hydroxyl group. Furthermore additional nitro groups have an additive influence if they are positioned in ortho or para locations. The trinitro compound shown at the lower right is a very strong acid called picric acid.
Comparing the Acidity of Alcohols with Phenols
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. Formulas illustrating this electron delocalization will be displayed when the "Resonance Structures" button beneath the previous diagram is clicked. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures.

The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.
Exercise
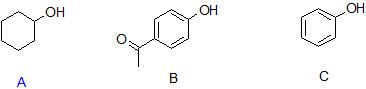
9. Arrange the following compounds in order of decreasing acidity when they are in solution.
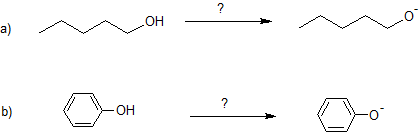
10. Specify the base needed to deprotonate each reactant.
- Answer
-
9. B > C > A
10. a) Na or NaH or NNH2
b) NaOH or KOH or LiOH
Contributors
Prof. Steven Farmer (Sonoma State University)
Jim Clark (Chemguide.co.uk)
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."