
11.4: Spectroscopy of Alcohols
- Page ID
- 182993
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Infrared Spectroscopy
If you look at an IR spectrum of 1-butanol, you will see:
- there are sp3 C-H stretching and CH2 bending modes at 2900 and 1500 cm-1.
- there is a strong C-O stretching mode near 1000 cm-1.
- there is a very large peak around 3400 cm-1. O-H peaks are usually very broad like this one.
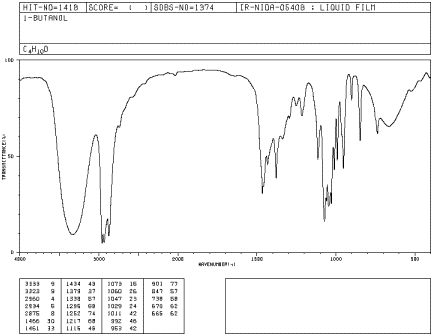
Peak shapes are sometimes very useful in recognizing what kind of bond is present. The rounded shape of most O-H stretching modes occurs because of hydrogen bonding between different hydroxy groups. Because protons are shared to varying extent with neighboring oxygens, the covalent O-H bonds in a sample of alcohol all vibrate at slightly different frequencies and show up at slightly different positions in the IR spectrum. Instead of seeing one sharp peak, you see a whole lot of them all smeared out into one broad blob. Since C-H bonds don't hydrogen bond very well, you don't see that phenomenon in an ether, and an O-H peak is very easy to distinguish in the IR spectrum.
Nuclear Magnetic Resonance Spectroscopy
Alcohols
Where is the -O-H peak? This is very confusing! Different sources quote totally different chemical shifts for the hydrogen atom in the -OH group in alcohols - often inconsistently. For example:

- The Nuffield Data Book quotes 2.0 - 4.0, but the Nuffield text book shows a peak at about 5.4.
- The OCR Data Sheet for use in their exams quotes 3.5 - 5.5.
- A reliable degree level organic chemistry text book quotes1.0 - 5.0, but then shows an NMR spectrum for ethanol with a peak at about 6.1.
- The SDBS database (used throughout this site) gives the -OH peak in ethanol at about 2.6.
The problem seems to be that the position of the -OH peak varies dramatically depending on the conditions - for example, what solvent is used, the concentration, and the purity of the alcohol - especially on whether or not it is totally dry.
A clever way of picking out the -OH peak
If you measure an NMR spectrum for an alcohol like ethanol, and then add a few drops of deuterium oxide, D2O, to the solution, allow it to settle and then re-measure the spectrum, the -OH peak disappears! By comparing the two spectra, you can tell immediately which peak was due to the -OH group.
The reason for the loss of the peak lies in the interaction between the deuterium oxide and the alcohol. All alcohols, such as ethanol, are very, very slightly acidic. The hydrogen on the -OH group transfers to one of the lone pairs on the oxygen of the water molecule. The fact that here we've got "heavy water" makes no difference to that.
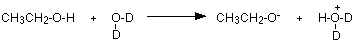
The negative ion formed is most likely to bump into a simple deuterium oxide molecule to regenerate the alcohol - except that now the -OH group has turned into an -OD group.
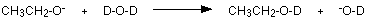
Deuterium atoms don't produce peaks in the same region of an NMR spectrum as ordinary hydrogen atoms, and so the peak disappears. You might wonder what happens to the positive ion in the first equation and the OD- in the second one. These get lost into the normal equilibrium which exists wherever you have water molecules - heavy or otherwise.
\[2D_2O \rightleftharpoons D_3O^+ + OH^-\]
The lack of splitting with -OH groups
Unless the alcohol is absolutely free of any water, the hydrogen on the -OH group and any hydrogens on the next door carbon don't interact to produce any splitting. The -OH peak is a singlet and you don't have to worry about its effect on the next door hydrogens.
The left-hand cluster of peaks is due to the CH2 group. It is a quartet because of the 3 hydrogens on the next door CH3 group. You can ignore the effect of the -OH hydrogen. Similarly, the -OH peak in the middle of the spectrum is a singlet. It hasn't turned into a triplet because of the influence of the CH2 group.
Mass Spectra Fragmentation Patterns
The fragmentation of molecular ions into an assortment of fragment ions is a mixed blessing. The nature of the fragments often provides a clue to the molecular structure, but if the molecular ion has a lifetime of less than a few microseconds it will not survive long enough to be observed. Without a molecular ion peak as a reference, the difficulty of interpreting a mass spectrum increases markedly. Fortunately, most organic compounds give mass spectra that include a molecular ion, and those that do not often respond successfully to the use of milder ionization conditions. Among simple organic compounds, the most stable molecular ions are those from aromatic rings, other conjugated pi-electron systems and cycloalkanes. Alcohols, ethers and highly branched alkanes generally show the greatest tendency toward fragmentation.
The presence of a functional group, particularly one having a heteroatom Y with non-bonding valence electrons (Y = N, O, S, X etc.), can dramatically alter the fragmentation pattern of a compound. This influence is thought to occur because of a "localization" of the radical cation component of the molecular ion on the heteroatom. After all, it is easier to remove (ionize) a non-bonding electron than one that is part of a covalent bond. By localizing the reactive moiety, certain fragmentation processes will be favored. These are summarized in the following diagram, where the green shaded box at the top displays examples of such "localized" molecular ions. The first two fragmentation paths lead to even-electron ions, and the elimination (path #3) gives an odd-electron ion. Note the use of different curved arrows to show single electron shifts compared with electron pair shifts.
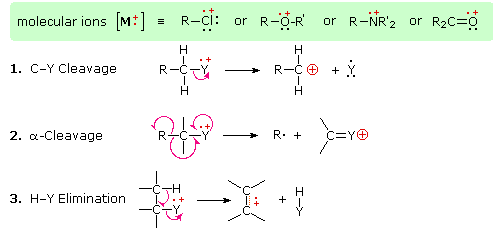
3-Pentanol shows three significant fragment ions. Alpha-fragmentation (loss of an ethyl radical) forms the m/z=59 base peak. Loss of water from this gives a m/z=41 fragment, and loss of ethene from m/z=59 gives a m/z=31 fragment.

Exercise
6. From mass spectroscopy analysis it was determined that a compound has the general formula C5H12O. Given the following 1H NMR spectrum, draw the structure. The integration values of each group of signals is given on the spectrum.
7. Given that alcohols are relatively acidic and the protons transfer in solution, what would you expect to happen to the NMR spectrum if D2O was used as a solvent.
8. From mass spectroscopy analysis it was determined that a compound has the general formula C3H8O. Given the following 1H NMR spectrum, draw the structure. The integration values of each group of signals is given on the spectrum.
- Answer
-
6.
7.
The alcohol proton signal's intensity in the 1H NMR would be expected to diminish and likely disappear. This is due to the fact that NMR can only probe the spin changes of nuclei with an odd number of protons.
8.
1-propanol
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry