
13.6: Collision Theory
- Page ID
- 168364

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Use the postulates of collision theory to explain the effects of physical state, temperature, and concentration on reaction rates
- Define the concepts of activation energy and transition state
- Use the Arrhenius equation in calculations relating rate constants to temperature
We should not be surprised that atoms, molecules, or ions must collide before they can react with each other. Atoms must be close together to form chemical bonds. This simple premise is the basis for a very powerful theory that explains many observations regarding chemical kinetics, including factors affecting reaction rates. Collision theory is based on the following postulates:
- The rate of a reaction is proportional to the rate of reactant collisions: \[\mathrm{reaction\: rate ∝ \dfrac{\#\,collisions}{time}} \nonumber \]
- The reacting species must collide in an orientation that allows contact between the atoms that will become bonded together in the product.
- The collision must occur with adequate energy to permit mutual penetration of the reacting species’ valence shells so that the electrons can rearrange and form new bonds (and new chemical species).
We can see the importance of the two physical factors noted in postulates 2 and 3, the orientation and energy of collisions, when we consider the reaction of carbon monoxide with oxygen:
\[\ce{2CO}(g)+\ce{O2}(g)⟶\ce{2CO2}(g) \nonumber \]
Carbon monoxide is a pollutant produced by the combustion of hydrocarbon fuels. To reduce this pollutant, automobiles have catalytic converters that use a catalyst to carry out this reaction. It is also a side reaction of the combustion of gunpowder that results in muzzle flash for many firearms. If carbon monoxide and oxygen are present in sufficient quantity, the reaction is spontaneous at high temperature and pressure.
The first step in the gas-phase reaction between carbon monoxide and oxygen is a collision between the two molecules:
\[\ce{CO}(g)+\ce{O2}(g)⟶\ce{CO2}(g)+\ce{O}(g) \nonumber \]
Although there are many different possible orientations the two molecules can have relative to each other, consider the two presented in Figure \(\PageIndex{1}\). In the first case, the oxygen side of the carbon monoxide molecule collides with the oxygen molecule. In the second case, the carbon side of the carbon monoxide molecule collides with the oxygen molecule. The second case is clearly more likely to result in the formation of carbon dioxide, which has a central carbon atom bonded to two oxygen atoms \(\ce{(O=C=O)}\). This is a rather simple example of how important the orientation of the collision is in terms of creating the desired product of the reaction.
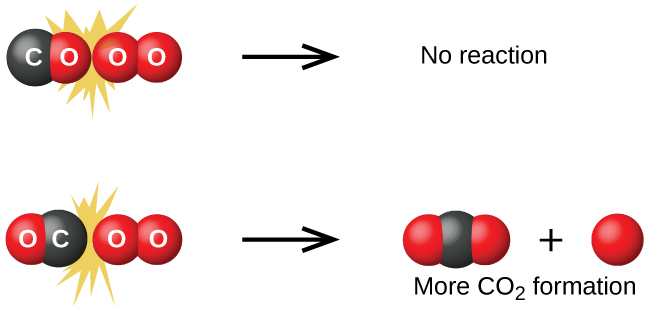
If the collision does take place with the correct orientation, there is still no guarantee that the reaction will proceed to form carbon dioxide. Every reaction requires a certain amount of activation energy for it to proceed in the forward direction, yielding an appropriate activated complex along the way. As Figure \(\PageIndex{2}\) demonstrates, even a collision with the correct orientation can fail to form the reaction product. In the study of reaction mechanisms, each of these three arrangements of atoms is called a proposed activated complex or transition state.
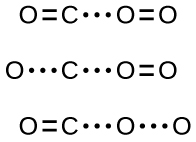
In most circumstances, it is impossible to isolate or identify a transition state or activated complex. In the reaction between carbon monoxide and oxygen to form carbon dioxide, activated complexes have only been observed spectroscopically in systems that utilize a heterogeneous catalyst. The gas-phase reaction occurs too rapidly to isolate any such chemical compound.
Collision theory explains why most reaction rates increase as concentrations increase. With an increase in the concentration of any reacting substance, the chances for collisions between molecules are increased because there are more molecules per unit of volume. More collisions mean a faster reaction rate, assuming the energy of the collisions is adequate.
Activation Energy and the Arrhenius Equation
The minimum energy necessary to form a product during a collision between reactants is called the activation energy (\(E_a\)). The kinetic energy of reactant molecules plays an important role in a reaction because the energy necessary to form a product is provided by a collision of a reactant molecule with another reactant molecule. (In single-reactant reactions, activation energy may be provided by a collision of the reactant molecule with the wall of the reaction vessel or with molecules of an inert contaminant.) If the activation energy is much larger than the average kinetic energy of the molecules, the reaction will occur slowly: Only a few fast-moving molecules will have enough energy to react. If the activation energy is much smaller than the average kinetic energy of the molecules, the fraction of molecules possessing the necessary kinetic energy will be large; most collisions between molecules will result in reaction, and the reaction will occur rapidly.
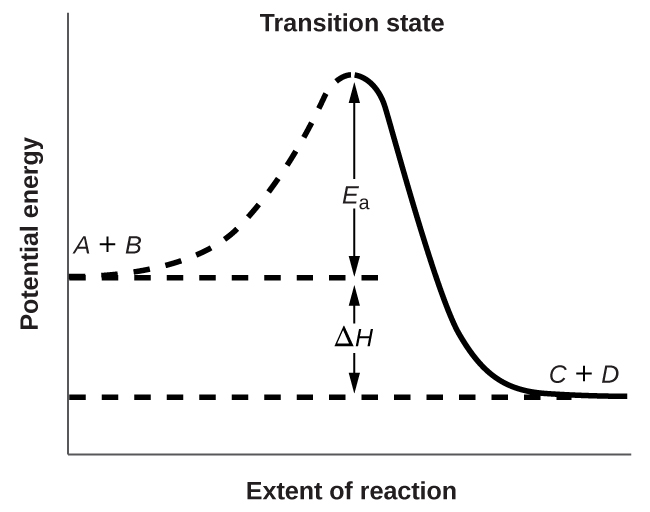
Figure \(\PageIndex{3}\) shows the energy relationships for the general reaction of a molecule of \(A\) with a molecule of \(B\) to form molecules of \(C\) and \(D\):
\[A+B⟶C+D \nonumber \]
The figure shows that the energy of the transition state is higher than that of the reactants \(A\) and \(B\) by an amount equal to \(E_a\), the activation energy. Thus, the sum of the kinetic energies of \(A\) and \(B\) must be equal to or greater than Ea to reach the transition state. After the transition state has been reached, and as \(C\) and \(D\) begin to form, the system loses energy until its total energy is lower than that of the initial mixture. This lost energy is transferred to other molecules, giving them enough energy to reach the transition state. The forward reaction (that between molecules \(A\) and \(B\)) therefore tends to take place readily once the reaction has started. In Figure \(\PageIndex{3}\), \(ΔH\) represents the difference in enthalpy between the reactants (\(A\) and \(B\)) and the products (\(C\) and \(D\)). The sum of \(E_a\) and \(ΔH\) represents the activation energy for the reverse reaction:
\[C+D⟶A+B \nonumber \]
We can use the Arrhenius equation to relate the activation energy and the rate constant, k, of a given reaction:
\[k=Ae^{−E_a/RT} \label{Arrhenius} \]
In this equation,
- \(R\) is the ideal gas constant, which has a value 8.314 J/mol/K,
- \(T\) is temperature on the Kelvin scale,
- \(E_a\) is the activation energy in joules per mole,
- \(e\) is the constant 2.7183, and
- \(A\) is a constant called the frequency factor, which is related to the frequency of collisions and the orientation of the reacting molecules.
Both postulates of the collision theory of reaction rates are accommodated in the Arrhenius equation. The frequency factor A is related to the rate at which collisions having the correct orientation occur. The exponential term, \(e^{−E_a/RT}\), is related to the fraction of collisions providing adequate energy to overcome the activation barrier of the reaction.
At one extreme, the system does not contain enough energy for collisions to overcome the activation barrier. In such cases, no reaction occurs. At the other extreme, the system has so much energy that every collision with the correct orientation can overcome the activation barrier, causing the reaction to proceed. In such cases, the reaction is nearly instantaneous.
The Arrhenius equation (Equation \ref{Arrhenius}) describes quantitatively much of what we have already discussed about reaction rates. For two reactions at the same temperature, the reaction with the higher activation energy has the lower rate constant and the slower rate. The larger value of \(E_a\) results in a smaller value for \(e^{−E_a/RT}\), reflecting the smaller fraction of molecules with enough energy to react. Alternatively, the reaction with the smaller \(E_a\) has a larger fraction of molecules with enough energy to react. This will be reflected as a larger value of \(e^{−E_a/RT}\), a larger rate constant, and a faster rate for the reaction.
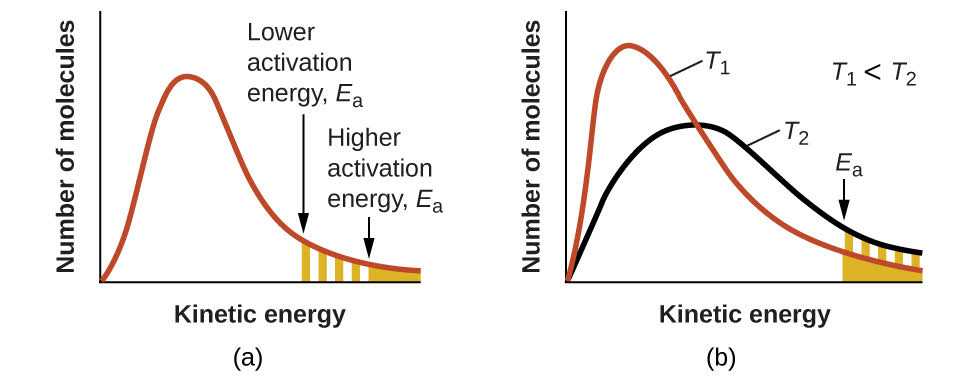
Figure \(\PageIndex{4}\): (a) As the activation energy of a reaction decreases, the number of molecules with at least this much energy increases, as shown by the shaded areas. (b) At a higher temperature, T2, more molecules have kinetic energies greater than Ea, as shown by the yellow shaded area.
An increase in temperature has the same effect as a decrease in activation energy. A larger fraction of molecules has the necessary energy to react (Figure \(\PageIndex{4}\)), as indicated by an increase in the value of \(e^{−E_a/RT}\). The rate constant is also directly proportional to the frequency factor, \(A\). Hence a change in conditions or reactants that increases the number of collisions with a favorable orientation for reaction results in an increase in \(A\) and, consequently, an increase in \(k\).
A convenient approach to determining \(E_a\) for a reaction involves the measurement of \(k\) at different temperatures and using of an alternate version of the Arrhenius equation that takes the form of linear equation:
\[\begin{align*}
\ln k&=\left(\dfrac{−E_a}{R}\right)\left(\dfrac{1}{T}\right)+\ln A\\
y&=mx+b
\end{align*} \nonumber \]
Thus, a plot of \(\ln k\) versus \(\dfrac{1}{T}\) gives a straight line with the slope \(\dfrac{-E_\ce{a}}{R}\), from which Ea may be determined. The intercept gives the value of \(\ln A\). This is sometimes call an Arrhenius Plot.
Determination of Ea The variation of the rate constant with temperature for the decomposition of HI(g) to H2(g) and I2(g) is given here. What is the activation energy for the reaction?
\[\ce{2HI}(g)⟶\ce{H2}(g)+\ce{I2}(g) \nonumber \]
| T (K) | k (L/mol/s) |
|---|---|
| 555 | 3.52 × 10−7 |
| 575 | 1.22 × 10−6 |
| 645 | 8.59 × 10−5 |
| 700 | 1.16 × 10−3 |
| 781 | 3.95 × 10−2 |
Solution
Values of \(\dfrac{1}{T}\) and ln k are:
| \(\mathrm{\dfrac{1}{T}\:(K^{−1})}\) | ln k |
|---|---|
| 1.80 × 10−3 | −14.860 |
| 1.74 × 10−3 | −13.617 |
| 1.55 × 10−3 | −9.362 |
| 1.43 × 10−3 | −6.759 |
| 1.28 × 10−3 | −3.231 |
Figure \(\PageIndex{5}\) is a graph of ln k versus \(\dfrac{1}{T}\). To determine the slope of the line, we need two values of ln k, which are determined from the line at two values of \(\dfrac{1}{T}\) (one near each end of the line is preferable). For example, the value of ln k determined from the line when \(\dfrac{1}{T}=1.25×10^{−3}\) is −2.593; the value when \(\dfrac{1}{T}=1.78×10^{−3}\) is −14.447.
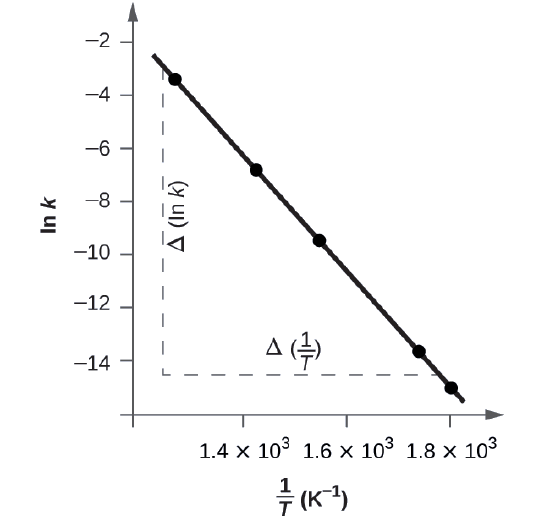
The slope of this line is given by the following expression:
\[\begin{align*}\ce{Slope}&=\dfrac{Δ(\ln k)}{Δ\left(\dfrac{1}{T}\right)}\\
&=\mathrm{\dfrac{(−14.447)−(−2.593)}{(1.78×10^{−3}\:K^{−1})−(1.25×10^{−3}\:K^{−1})}}\\
&=\mathrm{\dfrac{−11.854}{0.53×10^{−3}\:K^{−1}}=2.2×10^4\:K}\\
&=−\dfrac{E_\ce{a}}{R}
\end{align*} \nonumber \]
Thus:
\[ \begin{align*} E_\ce{a} &=\mathrm{−slope×\mathit R=−(−2.2×10^4\:K×8.314\: J\: mol^{−1}\:K^{−1})} \\[4pt] &=\mathrm{1.8×10^5\:J\: mol^{−1}} \end{align*} \nonumber \]
In many situations, it is possible to obtain a reasonable estimate of the activation energy without going through the entire process of constructing the Arrhenius plot. The Arrhenius equation:
\[\ln k=\left(\dfrac{−E_\ce{a}}{R}\right)\left(\dfrac{1}{T}\right)+\ln A \nonumber \]
can be rearranged as shown to give:
\[\dfrac{Δ(\ln k)}{Δ\left(\dfrac{1}{T}\right)}=−\dfrac{E_\ce{a}}{R} \nonumber \]
or
\[\ln\dfrac{k_1}{k_2}=\dfrac{E_\ce{a}}{R}\left(\dfrac{1}{T_2}−\dfrac{1}{T_1}\right) \nonumber \]
This equation can be rearranged to give a one-step calculation to obtain an estimate for the activation energy:
\[E_\ce{a}=−R\left( \dfrac{\ln k_2−\ln k_1}{\left(\dfrac{1}{T_2}\right)−\left(\dfrac{1}{T_1}\right)}\right ) \nonumber \]
Using the experimental data presented here, we can simply select two data entries. For this example, we select the first entry and the last entry:
| T (K) | k (L/mol/s) | \(\dfrac{1}{T}\:(K^{-1})\) | ln k |
|---|---|---|---|
| 555 | 3.52 × 10−7 | 1.80 × 10−3 | −14.860 |
| 781 | 3.95 × 10−2 | 1.28 × 10−3 | −3.231 |
After calculating \(\dfrac{1}{T}\) and ln k, we can substitute into the equation:
\[E_\ce{a}=\mathrm{−8.314\:J\:mol^{−1}\:K^{−1}\left(\dfrac{−3.231−(−14.860)}{1.28×10^{−3}\:K^{−1}−1.80×10^{−3}\:K^{−1}}\right)} \nonumber \]
and the result is Ea = 185,900 J/mol.
This method is very effective, especially when a limited number of temperature-dependent rate constants are available for the reaction of interest.
The rate constant for the rate of decomposition of N2O5 to NO and O2 in the gas phase is 1.66 L/mol/s at 650 K and 7.39 L/mol/s at 700 K:
\[\ce{2N2O5}(g)⟶\ce{4NO}(g)+\ce{3O2}(g) \nonumber \]
Assuming the kinetics of this reaction are consistent with the Arrhenius equation, calculate the activation energy for this decomposition.
- Answer
-
113,000 J/mol
Summary
Chemical reactions require collisions between reactant species. These reactant collisions must be of proper orientation and sufficient energy in order to result in product formation. Collision theory provides a simple but effective explanation for the effect of many experimental parameters on reaction rates. The Arrhenius equation describes the relation between a reaction’s rate constant and its activation energy, temperature, and dependence on collision orientation.
Key Equations
- \(k=Ae^{−E_a/RT}\)
- \(\ln k=\left(\dfrac{−E_\ce{a}}{R}\right)\left(\dfrac{1}{T}\right)+\ln A\)
- \(\ln\dfrac{k_1}{k_2}=\dfrac{E_\ce{a}}{R}\left(\dfrac{1}{T_2}−\dfrac{1}{T_1}\right)\)
Glossary
- activated complex
- (also, transition state) unstable combination of reactant species representing the highest energy state of a reaction system
- activation energy (Ea)
- energy necessary in order for a reaction to take place
- Arrhenius equation
- mathematical relationship between the rate constant and the activation energy of a reaction
- collision theory
- model that emphasizes the energy and orientation of molecular collisions to explain and predict reaction kinetics
- frequency factor (A)
- proportionality constant in the Arrhenius equation, related to the relative number of collisions having an orientation capable of leading to product formation