
14: Nuclear Magnetic Resonance
- Page ID
- 70785
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)
Nuclear magnetic resonance (NMR) is a versatile and highly-sophisticated spectroscopic technique which has been applied to a growing number of diverse applications in science, technology and medicine. This chapter will consider, for the most part, magnetic resonance involving protons.
Magnetic Properties of Nuclei
In all our previous work, it has been sufficient to treat nuclei as structureless point particles characterized fully by their mass and electric charge. On a more fundamental level, as was discussed in Chap. 1, nuclei are actually composite particles made of nucleons (protons and neutrons) and the nucleons themselves are made of quarks. The additional properties of nuclei which will now become relevant are their spin angular momenta and magnetic moments. Recall that electrons possess an intrinsic or spin angular momentum s which can have just two possible projections along an arbitrary spacial direction, namely \( \pm \frac{1}{2} \hbar \). Since \( \hbar \) is the fundamental quantum unit of angular momentum, the electron is classified as a particle of spin one-half. The electron’s spin state is described by the quantum numbers \( s=1\) and \( m_s = \pm 1 \). A circulating electric charge produces a magnetic moment \( \vec{\mu} \) proportional to the angular momentum J. Thus
\[ \vec{\mu} = \gamma \vec{J} \label{1}\]
where the constant of proportionality γ is known as the magnetogyric ratio. The z-component of \( \vec{\mu} \) has the possible values
\[\vec{\mu}_z = \gamma\hbar m_J \mbox{where} m_J = -J, -J+1,..., +J \label{2}\]
determined by space quantization of the angular momentum J. The energy of a magnetic dipole in a magnetic field B is given by
\[E= -\vec{\mu} \cdot \vec{B} = -\vec{\mu}_z B \label{3}\]
where magnetic field defines the z-axis. The SI unit of magnetic field (more correctly, magnetic induction) is the tesla, designated T. Electromagnets used in NMR produce fields in excess of 10 T. Small iron magnets have fields around .01 T, while some magnets containing rare-earth elements such as NIB (niobium-iron-boron) reach 0.2 T. The Earth’s magnetic field is approximately 5×10−5T (0.5 gauss in alternative units), dependent on geographic location. At the other extreme, a neutron star, which is really a giant nucleus, has a field predicted to be of the order of 108 T. The energy relation (3) determines the most conveniently units for magnetic moment, namely joules per tesla, J T−1.
For orbital motion of an electron, where the angular momentum is l, the magnetic moment is given by
\[ \vec{\mu}_z = -\frac{e{\hbar}}{2m} m_l = -\vec{\mu}_B m_l \label{4}\]
where the minus sign reflects the negative electric charge. The Bohr magneton is defined by
\[\vec{\mu}_B = -\frac{e{\hbar}}{2m} = 9.274\times10^{-24} JT^{-1} \label{5}\]
The magnetic moment produced by electron spin is written
\[\vec{\mu}_z = -g \vec{\mu}_B m_s \label{6}\]
with introduction of the g-factor. Eq (4) implies g = 1 for orbital motion. For electron spin, however, g = 2 (more exactly, 2.0023). The factor 2 compensates for ms = 1 such that spin and l = 1 orbital magnetic moments 2 are both equal to one Bohr magneton.
Many nuclei possess spin angular momentum, analogous to that of the electron. The nuclear spin, designated I, has an integral or half-integral value: 0, 1 , 1, 3 , and so on. Table 1 lists some nuclei of importance in chemical applications of NMR. The proton and the neutron both are spin 2 particles, like the electron. Complex nuclei have angular momenta which are resultants of the spins of their component nucleons. The deuteron 2H, with I = 1, evidently has parallel proton and neutron spins. The 4He nucleus has I = 0, as do 12C, 16O, 20Ne, 28Si and 32S. These nuclei contain filled shells of protons and neutrons with the vector sum of the component angular momenta equal to zero, analogous to closed shells of electrons in atoms and molecules. In fact, all even-even nuclei have spins of zero. Nuclear magnetic moments are of the order of a nuclear magneton
\[ \vec{\mu}_N = \frac{e\hbar}{2M} = 5.051\times 10^{-27} JT^{-1} \label{7}\]
where M is the mass of the proton. The nuclear magneton is smaller than the Bohr magneton by a factor m/M ≈ 1836.
Table 1: Some common nuclei in NMR spectroscopy

In analogy with Equations \(\ref{2}\) and \(\ref{6}\), nuclear moments are represented by
\[ \vec{\mu}_z = g_I \vec{\mu}_N m_I = \hbar \gamma_I m_I \label{8}\]
where gI is the nuclear g-factor and γI, the magnetogyric ratio. Most nuclei have positive g-factors, as would be expected for a rotating positive electric charge. It was long puzzling that the neutron, although lacking electric charge, has a magnetic moment. It is now understood that the neutron is a composite of three charged quarks, udd. The negatively-charged d- quarks are predominantly in the outermost regions of the neutron, thereby producing a negative magnetic moment, like that of the electron. The g- factor for 17O, and other nuclei dominated by unpaired neutron spins, is consequently also negative.
Nuclear Magnetic Resonance
The energy of a nuclear moment in a magnetic field, according to Equation \(\ref{3}\), is given by
\[ E_{m_I} = -\hbar \gamma_I m_I B \label{9}\]
For a nucleus of spin \(I\), the energy of a nucleus in a magnetic field is split into \(2I+1\) Zeeman levels. A proton and other nuclei with spin \( \frac{1}{2}\) particles will have just two possible levels:
\[E_{\pm \frac{1}{2}} = \pm \dfrac{1}{2} \hbar \gamma B \label{10}\]
with the α-spin state ( \( m_I = -\frac{1}{2} \)) lower in energy than the β-spin state (\( m_I = +\frac{1}{2} \)) by
\[\Delta E = \hbar \gamma B \label{11}\]
Fig. 1 shows the energy of a proton as a function of magnetic field. In zero field (B = 0), the two spin states are degenerate. In a field B, the energy splitting corresponds to a photon of energy \(\Delta E = \hbar \omega = h \nu \) where
\[ \omega_L = \gamma_B\space \mbox{or}\space \nu_L = \gamma_B \label{12}\]
known as the Larmor frequency of the nucleus. For the proton in a field of 1 T, \(\nu_L \) = 42.576 MHz, as the proton spin orientation flips from \( +\frac{1}{2} \) to \( -\frac{1}{2} \). This transition is in the radiofrequency region of the electromagnetic spectrum. NMR spectroscopy consequently exploits the technology of radiowave engineering.
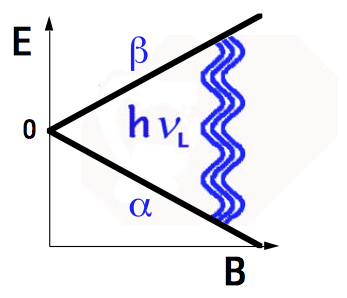
Figure 1. Energies of spin \( \frac{1}{2} \) in magnetic field showing NMR transition at Larmor frequency \( \nu_L \).
A transition cannot occur unless the values of the radiofrequency and the magnetic field accurately fulfill Eq (12). This is why the technique is categorized as a resonance phenomenon. If some resonance condition is not satisfied, no radiation can be absorbed or emitted by the nuclear spins. In the earlier techniques of NMR spectroscopy, it was found more convenient keep the radiofrequency fixed and sweep over values of the magnetic field B to detect resonances. These have been largely supplanted by modern pulse techniques, to be described later.
The transition probability for the upward transition (absorption) is equal to that for the downward transition (stimulated emission). (The contribution of spontaneous emission is neglible at radiofrequencies.) Thus if there were equal populations of nuclei in the α and β spin states, there would be zero net absorption by a macroscopic sample. The possibility of observable NMR absorption depends on the lower state having at least a slight excess in population. At thermal equilibrium, the ratio of populations follows a Boltzmann distribution
\[ \frac{N_{\beta}}{N_\alpha} = \frac{e^{\frac{-E_\beta}{kT}}}{e^{\frac{-E_\alpha}{kt}}} = e^{- \frac{\hbar \gamma B}{kT}} \label{13}\]
Thus the relative population difference is given by
\[ \dfrac{\Delta N}{N_\alpha} = \frac{N_\alpha - N_{\beta}}{N_\alpha + N_{\beta}} \approx \frac{ \hbar \gamma B}{2kT} \label{14}\]
Since nuclear Zeeman energies are so small, the populations of the α and β spin states differ very slightly. For protons in a 1 T field, ∆N/N ≈ 3×10−6. Although the population excess in the lower level is only of the order of parts per million, NMR spectroscopy is capable of detecting these weak signals. Higher magnetic fields and lower temperatures are favorable conditions for enhanced NMR sensitivity.
The Chemical Shift
NMR has become such an invaluable technique for studying the structure of atoms and molecules because nuclei represent ideal noninvasive probes of their electronic environment. If all nuclei of a given species responded at their characteristic Larmor frequencies, NMR might then be useful for chemical analysis, but little else. The real value of NMR to chemistry comes from minute differences in resonance frequencies dependent on details of the electronic structure around a nucleus. The magnetic field induces orbital angular momentum in the electron cloud around a nucleus, thus, in effect, partially shielding the nucleus from the external field B. The actual or local value of the magnetic field at the position of a nucleus is expressed
\[ B_{loc} = (1- \sigma)B \label{15}\]
where the fractional reduction of the field is denoted by σ, the shielding constant, typically of the order of parts per million. The actual resonance frequency of the nucleus in its local environment is then equal to
\[ \nu = (1- \sigma) \frac{\gamma B}{2 \pi} \label{16}\]
A classic example of this effect is the proton NMR spectrum of ethanol CH3CH2OH, shown in Fig. 2. The three peaks, with intensity ratios 3:2:1 can be identified with the three chemically-distinct environments in which the protons find themselves: three methyl protons (CH3), two methylene protons (CH2) and one hydroxyl proton (OH).
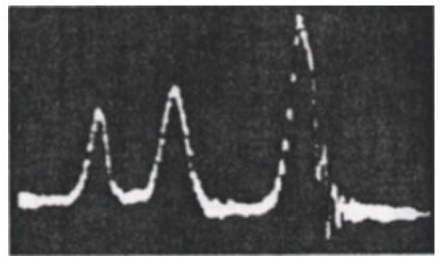
Figure 2. Oscilloscope trace showing the first NMR spectrum of ethanol, taken at Stanford University in 1951. Courtesy Varian Associates, Inc.
The variation in resonance frequency due to the electronic environment of a nucleus is called the chemical shift. Chemical shifts on the delta scale are defined by
\[ \delta = {\frac{ \nu - \nu^{O}}{\nu^{O}}} \times 10^{6} \label{17}\]
where νo represents the resonance frequency of a reference compound, usually tetramethylsilane Si(CH3)4, which is rich in highly-shielded chemically-equivalent protons, as well as being unreactive and soluble in many liquids. By definition δ = 0 for TMS and almost everything else is “downfield” with positive values of δ. Most compounds have delta values in the range of 0 to 12 (hydrogen halides have negative values, e.g. δ ≈ −13 for HI). The hydrogen atom has δ ≈ 13 while the bare proton would have δ ≈ 31. Conventionally, the δ-scale is plotted as increasing from right to left, in the opposite sense to the magnitude of the magnetic field. Nuclei with larger values of δ are said to be more deshielded, with the bare proton being the ultimate limit. Fig. 3 shows some typical values of δ for protons in some common organic compounds.
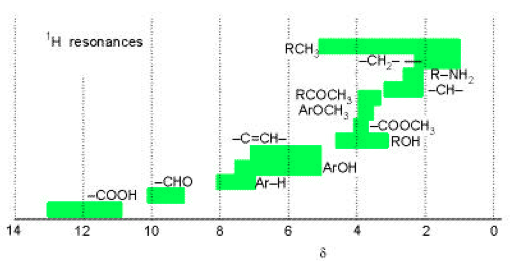
Figure 3. Ranges of proton chemical shifts for common functional groups. From P. Atkins, Physical Chemistry, (Freeman, New York, 2002).
Fig. 4 shows a high-resolution NMR spectrum of ethanol, including a δ-scale. The “fine structure” splittings of the three chemically-shifted components will be explained in the next Section. The chemical shift of a nucleus is very difficult to treat theoretically. However, certain empirical regularities, for example those represented in Fig. 3, provide clues about the chemical environment of the nucleus. We will not consider these in any detail except to remark that often increased deshielding of a nucleus (larger δ) can often be attributed to a more electronegative neighboring atom. For example the proton in the ethanol spectrum (Fig. 4) with δ ≈ 5 can be identified as the hydroxyl proton, since the oxygen atom can draw significant electron density from around the proton.
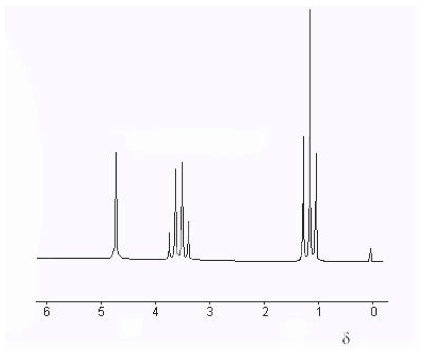
Figure 4. High-resolution NMR spectrum of ethanol showing δ scale of chemical shifts. The line at δ = 0 corresponds to the TMS trace added as a reference.
Neighboring groups can also contribute to the chemical shift of a given atom, particularly those with mobile π-electrons. For example, the ring current in a benzene ring acts as a secondary source of magnetic field. Depending on the location of a nucleus, this can contribute either shielding or deshielding of the external magnetic field, as shown in Fig. 5. The interaction of neighboring groups can be exploited to obtain structural information by using lanthanide shift reagents. Lanthanides (elements 58 through 71) contain 4f-electrons, which are not usually involved in chemical bonding and can give large paramagnetic contributions. Lanthanide complexes which bind to organic molecules can thereby spread out pro- ton resonances to simplify their analysis. A popular chelating complex is Eu(dpm)3, tris(dipivaloylmethanato)europium, where dpm is the group (CH3)3C–CO=CH–CO–C(CH3)3.
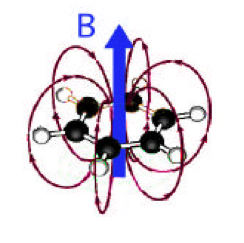
Figure 5. Magnetic field produced by ring current in benzene, shown as red loops. Where the arrows are parallel to the external field B, including protons directly attached to the ring, the effect is deshielding. However, any nuclei located within the return loops will experience a shielding effect.
Spin-Spin Coupling
Two of the resonances in the ethanol spectrum shown in Fig. 4 are split into closely-spaced multiplets—one triplet and one quartet. These are the result of spin-spin coupling between magnetic nuclei which are relatively close to one another, say within two or three bond separations. Identical nuclei in identical chemical environments are said to be equivalent. They have equal chemical shifts and do not exhibit spin-spin splitting. Nonequivalent magnetic nuclei, on the other hand, can interact and thereby affect one another’s NMR frequencies. A simple example is the HD molecule, in which the spin-1proton can interact with the spin-1 deuteron, even though the atoms are chemically equivalent. The proton’s energy is split into two levels by the external magnetic field, as shown in Fig. 1. The neighboring deuteron, itself a magnet, will also contribute to the local field at the pro- ton. The deuteron’s three possible orientations in the external field, with MI = −1,0,+1, with different contributions to the magnetic field at the proton, as shown in Fig. 6. The proton’s resonance is split into three evenly spaced, equally intense lines (a triplet), with a separation of 42.9 Hz. Corespondingly the deuteron’s resonance is split into a 42.9 Hz doublet by its interaction with the proton. These splittings are independent of the external field B, whereas chemical shifts are proportional to B. Fig. 6 represents the energy levels and NMR transitions for the proton in HD.

-
Figure 6. Nuclear energy levels for proton in HD molecule. The two Zeeman levels of the proton when B > 0 are further split by interaction with the three possible spin orientations of the deuteron Md= −1, 0, +1. The proton NMR transition, represented by blue arrows, is split into a triplet with separation 42.9 Hz.
Nuclear-spin phenomena in the HD molecule can be compactly represented by a spin Hamiltonian
\[ \hat{H} = -\hbar \gamma_H M_H(1- \sigma_H) - \hbar \gamma_D M_D(1-\sigma_D)B + h J_{HD} I_H \cdot I_D \label{18}\]
-
The shielding constants σH and σD are, in this case, equal since the two nuclei are chemically identical. For sufficiently large magnetic fields B, the last term is effectively equal to hJHDMHMD. The spin-coupling constant J can be directly equated to the splitting expressed in Hz. We consider next the case of two equivalent protons, for example, the CH2 group of ethanol. Each proton can have two possible spin states with \( M_I = \pm\frac{1}{2} \) , giving a total of four composite spin states. Just as in the case of 2 electron spins, these combine to give singlet and triplet nuclear-spin states with M = 0 and 1, respectively. Also, just as for electron spins, transitions between singlet and triplet states are forbidden. The triplet state allows NMR transitions with \( \Delta M = \pm 1 \) to give a single resonance frequency, while the singlet state is inactive. As a consequence, spin-spin splittings do not occur among identical nuclei. For example, the H2 molecule shows just a single NMR frequency. And the CH2 protons in ethanol do not show spin- spin interactions with one another. They can however cause a splitting of the neighboring CH3 protons. Fig. 7 (left side) shows the four possible spin states of two equivalent protons, such as those in the methylene group CH2, and the triplet with intensity ratios 1:2:1 which these produce in nearby protons. Also shown (right side) are the eight possible spin states for three equivalent protons, say those in a methyl group CH3, and the quartet with intensity ratios 1:3:3:1 which these produce. In general, n equivalent protons will give a splitting pattern of n + 1 lines in the ratio of binomial coefficients 1:n:n(n − 1)/2 . . . The tertiary hydrogen in isobutane (CH3)3CH∗, marked with an asterisk, should be split into 10 lines by the 9 equivalent methyl protons.
-
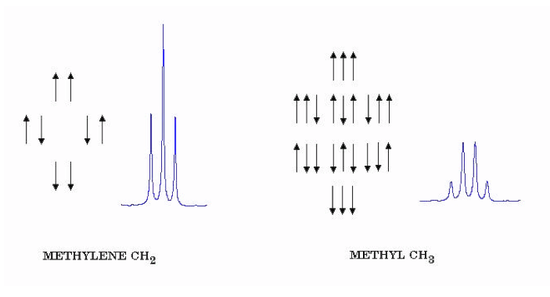
Figure 7. Splitting patterns from methylene and methyl protons.
The NMR spectrum of ethanol CH3CH2OH (Fig. 4) can now be interpreted. The CH3 protons are split into a 1:2:1 triplet by spin-spin interaction with the neighboring CH2. Conversely, the CH2 protons are split into a 1:3:3:1 quartet by interaction with the CH3. The OH (hydroxyl) proton evidently does not either cause or undergo spin-spin splitting. The explanation for this is hydrogen bonding, which involves rapid exchange of hydroxyl protons among neighboring molecules. If this rate of exchange is greater than or comparable to the NMR radiofrequency, then the splittings will be “washed out.” Only one line with a motion-averaged value of the chemical shift will be observed. NMR has consequently become a useful tool to study intramolecular motions.
Contributors and Attributions
Seymour Blinder (Professor Emeritus of Chemistry and Physics at the University of Michigan, Ann Arbor)