
9: Atomic Structure and The Periodic Law
- Page ID
- 70780
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)
Quantum mechanics can account for the periodic structure of the elements, by any measure a major conceptual accomplishment for any theory. Although accurate computations become increasingly more challenging as the number of electrons increases, the general patterns of atomic behavior can be predicted with remarkable accuracy.
Slater Determinants
According to the orbital approximation, which was introduced in the last Chapter, an N-electron atom contains N occupied spinorbitals, which can be designated \(\phi\)a, \(\phi\)b . . . \(\phi\)n. In accordance with the Pauli exclusion principle, no two of these spinorbitals can be identical. Also, every electron should be equally associated with every spinorbital. A very neat mathematical representation for these properties is a generalization of the two-electron wavefunction (8.13) or (8.15) called a Slater determinant
\[ \Psi (1,2 \ldots N ) = \frac{1}{\sqrt{N !}} \begin{vmatrix} \phi_a(1) & \phi_b(1) & \ldots & \phi_n(1) \\ \phi_a(2) & \phi_b(2) & \ldots & \phi_n(2) \\ & & \vdots & \\ \phi_a(N) & \phi_b(N) & \ldots & \phi_n(N) \\ \end{vmatrix} \label{1} \]
Since interchanging any two rows (or columns) of a determinant multiplies it by \(-1\), the antisymmetry property (8.15) is fulfilled, for every pair of electrons.
The Hamiltonian for an atom with N electrons around a nucleus of charge Z can be written
\[ \hat{H} = \sum_{i=1}^N \left\{-\frac{1}{2}\bigtriangledown^2_i - \frac{Z}{r_i} \right\} + \sum_{i<j}^N \frac{1}{ r_{ij}}\label{2} \]
The sum over electron repulsions is written so that each pair {i,,j} is counted just once. The energy of the state represented by a Slater determinant (Equation \(\ref{1}\)) can be obtained after a lengthy derivation. We give just the final result
\[ \tilde{E} = \sum_{a} I_a+\frac{1}{2}\sum_{a,b} \left( J_{ab}-K_{ab} \right) \label{3} \]
where the sums run over all occupied spinorbitals. The one-electron, Coulomb and exchange integrals have the same form as those defined for helium atom in Eqs (8.22-24). The only difference is that an exchange integral equals zero unless the spins of orbitals a and b are both \(\alpha\) or both \(\beta\). The factor 1/2 corrects for the double counting of pairs of spinorbitals in the second sum. The contributions with a = b can be omitted since Jaa = Kaa. This effectively removes the Coulomb interaction of an orbital with itself, which is spurious.
The Hartree-Fock or self-consistent field (SCF) method is a procedure for optimizing the orbital functions in the Slater determinant (1), so as to minimize the energy (Equation \(\ref{3}\)). SCF computations have been carried out for all the atoms of the periodic table, with predictions of total energies and ionization energies generally accurate in the \(1-2\%\) range.
Aufbau Principles and Periodic Structure
Aufbau means "building-up." Aufbau principles determine the order in which atomic orbitals are filled as the atomic number is increased. For the hydrogen atom, the order of increasing orbital energy is given by 1s < 2s = 2p < 3s = 3p = 3d, etc. The dependence of energy on n alone leads to extensive degeneracy, which is however removed for orbitals in many-electron atoms. Thus 2s lies below 2p, as already observed in helium. Similarly, 3s, 3p and 3d increase energy in that order, and so on. The 4s is lowered sufficiently that it becomes comparable to 3d. The general ordering of atomic orbitals is summarized in the following scheme:
\[ 1s < 2s < 2p < 3s < 3p < 4s \sim 3d < 4p < 5s \sim 4d\\< 5p < 6s \sim 5d \sim 4f < 6p < 7s \sim 6d \sim 5f \label{4}\]
and illustrated in Figure 1. This provides enough orbitals to fill the ground states of all the atoms in the periodic table. For orbitals designated as comparable in energy, e.g., 4s \(\sim\) 3d, the actual order depends which other orbitals are occupied. The sequence of orbitals pictured above increases in the order \(n+\frac{1}{2}\)l, except that l = 4 (rather than 3) is used for an f-orbital.

The tabulation below shows the ground-state electron configuration and term symbol for selected elements in the first part of the periodic table. From the term symbol, one can read off the total orbital angular momentum L and the total spin angular momentum S. The code for the total orbital angular momentum mirrors the one-electron notation, but using upper-case letters, as follows:
L = 0 1 2 3 4
S P D F G
The total spin S is designated, somewhat indirectly, by the spin multiplicity 2S + 1 written as a superscript before the S, P, D. . . symbol. For example 1S (singlet S) ,1P (singlet P). . . mean S = 0; 2S (doublet S) ,2P (doublet P). . . mean S = 1/2; 3S (triplet S) ,3P (triplet P). . . mean S = 1, and so on. Please do not confuse the spin quantum number S with the orbital designation S.
| Atom | Z | Electron Configuration | Term Symbol |
|---|---|---|---|
| H | 1 | 1s | 2S1/2 |
| He | 2 | 1s2 | 1S0 |
| Li | 3 | [He]2s | 2S1/2 |
| Be | 4 | [He]2s2 | 1S0 |
| B | 5 | [He]2s22p | 2P1/2 |
| C | 6 | [He]2s22p2 | 3P0 |
| N | 7 | [He]2s22p3 | 4S3/2 |
| O | 8 | [He]2s22p4 | 3P2 |
| F | 9 | [He]2s22p5 | 2P3/2 |
| Ne | 10 | [He]2s22p6 | 1S0 |
| Na | 11 | [Ne]3s | 2S1/2 |
| Cl | 17 | [Ne]3s23p5 | 2P3/2 |
| Ar | 18 | [Ne]3s23p6 | 1S0 |
| K | 19 | [Ar]4s | 2S1/2 |
| Ca | 20 | [Ar]4s2 | 1S0 |
| Sc | 21 | [Ar]4s23d | 2D3/2 |
| Ti | 22 | [Ar]4s23d2 | 3F2 |
| V | 23 | [Ar]4s23d3 | 4F3/2 |
| Cr | 24 | [Ar]4s3d5 | 7S3 |
| Mn | 25 | [Ar]4s23d5 | 6S5/2 |
| Fe | 26 | [Ar]4s23d6 | 5D4 |
| Co | 27 | [Ar]4s23d7 | 4F9/2 |
| Ni | 28 | [Ar]4s23d8 | 3F4 |
| Cu | 29 | [Ar]4s3d10 | 2S1/2 |
| Zn | 30 | [Ar]4s23d10 | 1S0 |
| Ga | 31 | [Ar]4s23d104p | 2P1/2 |
| Br | 35 | [Ar]4s23d104p5 | 2P3/2 |
| Kr | 36 | [Ar]3d104s24p6 | 1S0 |
The vector sum of the orbital and spin angular momentum is designated
\[ \bf{J} = \bf{L} + \bf{S} \label{5} \]
The possible values of the total angular momentum quantum number J runs in integer steps between |L - S| and L + S. The J value is appended as a subscript on the term symbol, eg, 1S0, 2P1/2, 2P3/2. The energy differences between J states is a result of spin-orbit interaction, a magnetic interaction between the circulating charges associated with orbital and spin angular momenta. For atoms of low atomic number, the spin-orbit coupling is a relatively small correction to the energy, but it can become increasingly significant for heavier atoms.
We will next consider in some detail the Aufbau of ground electronic states starting at the beginning of the periodic table. Hydrogen has one electron in an s-orbital so its total orbital angular momentum is also designated S. The single electron has s = 1/2, thus S = 1/2. The spin multiplicity 2S + 1 equals 2, thus the term symbol is written 2S. In helium, a second electron can occupy the 1s shell, provided it has the opposite spin. The total spin angular momentum is therefore zero, as is the total orbital angular momentum. The term symbol is 1S, as it will be for all other atoms with complete electron shells. In determining the total spin and orbital angular moments, we need consider only electrons outside of closed shells. Therefore lithium and beryllium are a reprise of hydrogen and helium. The angular momentum of boron comes from the single 2p electron, with l = 1 and s = 1/2, giving a 2P state.
To build the carbon atom, we add a second 2p electron. Since there are three degenerate 2p orbitals, the second electron can go into either the already-occupied 2p orbital or one of the unoccupied 2p orbitals. Clearly, two electrons in different 2p orbitals will have less repulsive energy than two electrons crowded into the same 2p orbital. In terms of the Coulomb integrals, we would expect, for example
\[ J(2px, 2py) < J(2px, 2px) \label{6} \]
For nitrogen atom, with three 2p electrons, we expect, by the same line of reasoning, that the third electron will go into the remaining unoccupied 2p orbital. The half-filled 2p3 subshell has an interesting property. If the three occupied orbitals are 2px, 2py and 2pz, then their total electron density is given by
\[ \rho_{2p} = \psi^{2}_{2p_{x}} + \psi^{2}_{2p_{y}} + \psi^{2}_{2p_{z}} = \left(x^2 + y^2 + z^2\right) \times \text{function of r} = \text{function of r} \label{7} \]
noting that \( x^2 + y^2 + z^2 = r^2 \). But spherical symmetry implies zero angular momentum, like an s-orbital. In fact, any half filled subshell, such as p3, d5, f7, will contribute zero angular momentum. The same is, of course true as well for filled subshells, such as p6, d10, f14. These are all S terms.
Another way to understand this vector cancelation of angular momentum is to consider the alternative representation of the degenerate 2p-orbitals: 2p-1; 2p0 and 2p1. Obviously, the z-components of angular momentum now add to zero, and since only this one component is observable, the total angular momentum must also be zero.
Returning to our unfinished consideration of carbon, the 2p2 subshell can be regarded, in concept, as a half-filled 2p3 subshell plus an electron "hole." The advantage of this picture is that the total orbital angular momentum must be equal to that of the hole, namely l = 1. This is shown below:
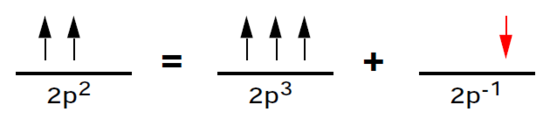
Thus the term symbol for the carbon ground state is P. It remains to determine the total spins of these subshells. Recall that exchange integrals Kab are non-zero only if the orbitals a and b have the same spin. Since exchange integrals enter the energy formula (3) with negative signs, the more nonvanishing K integrals, the lower the energy. This is achieved by having the maximum possible number of electrons with unpaired spins. We conclude that S = 1 for carbon and S = 3/2 for nitrogen, so that the complete term symbols are 3P and 4S, respectively.
The allocation electrons among degenerate orbitals can be formalized by Hund's rule: For an atom in its ground state, the term with the highest multiplicity has the lowest energy.
Resuming Aufbau of the periodic table, oxygen with four 2p electrons must have one of the 2p-orbitals doubly occupied. But the remaining two electrons will choose unoccupied orbitals with parallel spins. Thus oxygen has, like carbon, a 3P ground state. Fluorine can be regarded as a complete shell with an electron hole, thus a 2P ground state. Neon completes the 2s2p shells, thus term symbol 1S. The chemical stability and high ionization energy of all the noble-gas atoms can be attributed to their electronic structure of complete shells. The third row of the periodic table is filled in complete analogy with the second row. The similarity of the outermost electron shells accounts for the periodicity of chemical properties. Thus, the alkali metals Na and K belong in the same family as Li, the halogens Cl and Br are chemically similar to F, and so forth.
The transition elements, atomic numbers 21 to 30, present further challenges to our understanding of electronic structure. A complicating factor is that the energies of the 4s and 3d orbitals are very close, so that interactions among occupied orbitals often determines the electronic state. Ground-state electron configurations can be deduced from spectroscopic and chemical evidence, and confirmed by accurate self-consisent field computations. The 4s orbital is the first to be filled in K and Ca. Then come 3d electrons in Sc, Ti and V. A discontinuity occurs at Cr. The groundstate configuration is found to be 4s3d5, instead of the extrapolated 4s23d4. This can be attributed to the enhanced stability of a half-filled 3d5-shell. All six electrons in the valence shells have parallel spins, maximizing the number of stabilizing exchange integrals and giving the observed 6S term. An analogous discontinuity occurs for copper, in which the 4s subshell is again raided to complete the 3d10 subshell.
The order in which orbitals are filled is not necessarily consistent with the order in which electrons are removed. Thus, in all the positive ions of transition metals, the two 4s-electrons are removed first. The inadequacy of any simple generalizations about orbital energies is demonstrated by comparing the three ground-state electron configurations: Ni 4s23d8, Pd 5s04d10 and Pt 6s5d9.
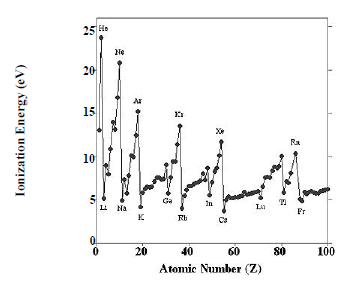
The periodic structure of the elements is evident for many physical and chemical properties, including chemical valence, atomic radius, electronegativity, melting point, density, and hardness. The classic prototype for periodic behavior is the variation of the first ionization energy with atomic number, which is plotted in in Figure 2.
Contributors and Attributions
Seymour Blinder (Professor Emeritus of Chemistry and Physics at the University of Michigan, Ann Arbor)
- Caitlin Kozack (Hope College)