4.4: Applications of Symmetry in Chemistry
- Page ID
- 326175
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In the previous sections of this chapter, the necessary tools to assign a point group to a molecule and to be able to read a character table were introduced. A good question at this point is: What is the importance of these tools in inorganic chemistry?
In fact, there are many applications in the study of inorganic chemistry where these symmetry tools become handy. In general, these applications belong to one of the following categories:
- Determining orbital overlap for molecular orbitals.
- Understanding the spectroscopic properties of molecules
- Identifying chiral molecular species
Orbital Overlap and Molecular Orbitals
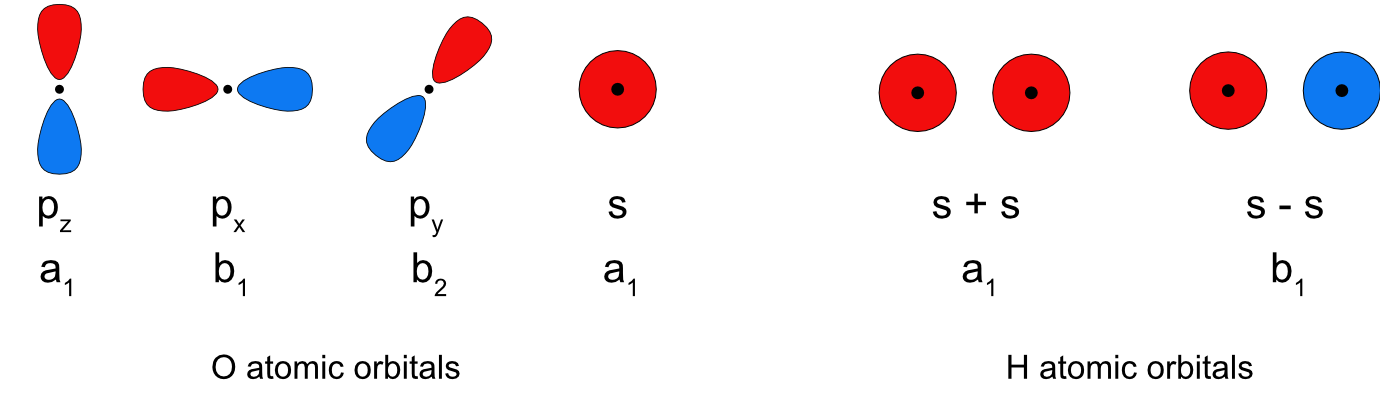
As discussed previously, symmetry is one of the criteria for determining whether two or more atomic orbitals can overlap to form molecular orbitals. Once molecules get larger than two or three atoms drawing all the possible atomic orbital combinations to determine if they have the right symmetry to overlap is challenging and time consuming. Symmetry can be applied in this case to make the determination more quickly and easily, only orbitals that have the same irreducible representation have the correct symmetry to overlap and form a molecular orbital. As an example we will revisit the orbital overlap in water. Following the flow chart, the water molecule is in the C2v point group because it has a C2 principal rotation axis, no other rotation axes, no σh reflection plane and 2 σv reflection planes. Figure \(\PageIndex{1}\) shows the valence atomic orbitals of oxygen and hydrogen along with the irreducible representations they correspond to in the C2v point group. Because only orbitals with the same irreducible representation can form molecular orbitals, both the 2pz and 2s on O can overlap with the in-phase combination of 1s orbitals on the H atoms and the 2px on O can overlap with the out of phase combination of 1s orbitals on the H atoms. However, the 2py orbital on O has a different irreducible representation from both the H 1s combinations. This means the O 2py is non bonding in water.
Vibrational Spectroscopy
Every molecule has a total of \(3n-6\) (\(3n-5\) for linear) vibrational modes, but not all of those vibrations will necessarily appear in the infrared or Raman spectrum of the molecule. Symmetry can be used to develop selection rules, and easily determine which vibrational modes will be infrared active, which will be Raman active, and which will be both.
(a)
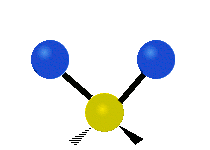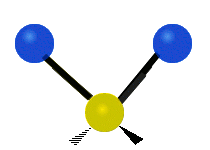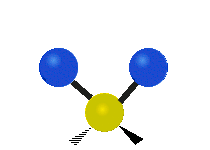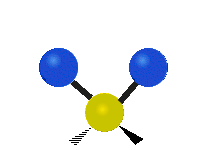
(b)
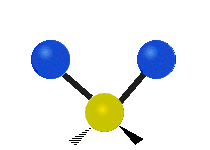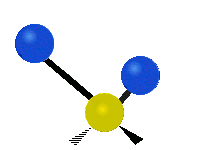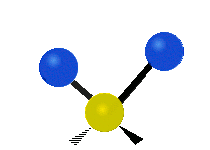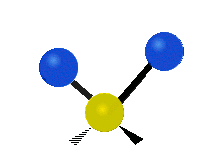
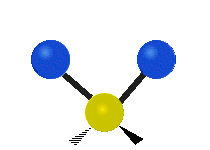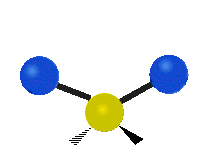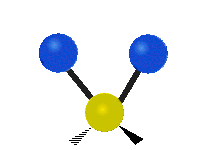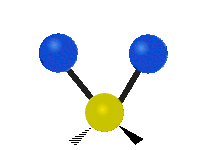
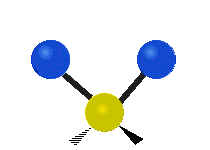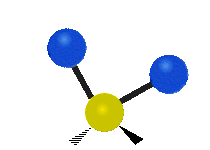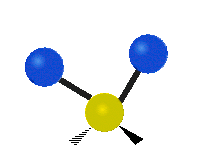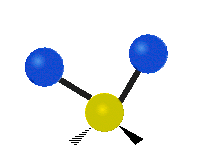
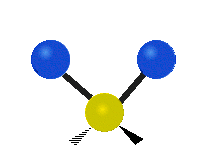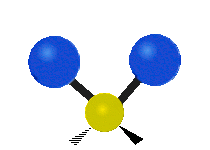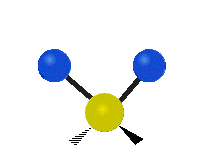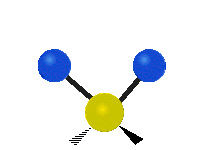
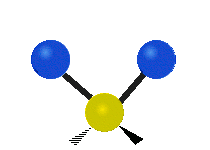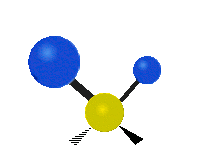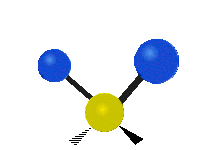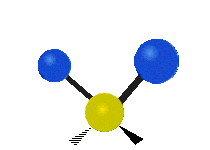
Selection Rules
Infrared Spectroscopy
Infrared spectroscopy measures the frequency of absorption when a sample is irradiated with infrared electromagnetic radiation. The bonds between atoms can be thought of as a spring connecting two masses. In the spring-mass analogy the moving system can be approximated by a simple harmonic oscillator. The frequency oscillation is proportional to \({\displaystyle {\sqrt {\frac {k}{m}}}}\),where k is the spring constant and m is the mass of the object. In a similar approximation, the frequency of vibration between two atoms is unique and varies depending on the strength of the bond (k) and the size of the atoms (m). When the frequency of electromagnetic radiation matches the frequency of vibration between atoms, the energy can be possibly be absorbed. In order for a vibrational mode to be infrared active (absorb infrared light) the vibration must produce a change in the dipole moment of the molecule which can interact with the electric field. Any vibrational mode which has the same symmetry (corresponds to the same irreducible representation) as the mathematical functions \(x\), \(y\), and \(z\) will shift the charge distribution of the molecule in any of the x, y, or z directions resulting in a change in the dipole moment. Any vibrational mode which has the same symmetry as \(x\), \(y\), or \(z\) will be able to absorb infrared light and will appear in an infrared spectrum.
Raman Spectroscopy
Unlike IR spectroscopy which measures the energy absorbed, Raman spectroscopy is based on inelastic scattering of visible light. Upon a collision between a molecule and photon, a small amount of energy, equal to a vibrational frequency, can be transferred to or from the photon. The difference in the photon's energy before and after the collision is the Raman shift, and will have the same energies as the (Raman active) vibrational frequencies of the molecule. A vibrational mode will be Raman active if it causes a change in the molecular polarizability. Polarizability measures the ability for a molecule’s electron cloud to become distorted. A dense electron cloud is more difficult to change than a more diffuse electron density. Any vibrational mode which has the same symmetry (corresponds to the same irreducible representation) as the product mathematical functions will cause a change in polarizability and will be Raman active. Product functions include any of the functions: \(xy, xz, yz, x2, y2, z2\), or any combination, and are listed in the second column of math functions in a character table.
Rule of Mutual Exclusion
A molecule is centrosymmetric if it has a center of inversion and their corresponding point group contains the class for inversion. In such cases, the functions \(x\), \(y\), and \(z\) belong to ungerade (odd) irreducible representations, and the product functions belong to gerade (even) irreducible representations. As a result, vibrational modes in centrosymmetric molecules may be either infrared or Raman active, but they cannot be both.
Example \(\PageIndex{1}\)
The bond stretching vibrations in [PtCl4]2- are shown below, along with their corresponding irreducible representations. [PtCl4]2- is a square planar molecule in the D4h point group.
 Figure \(\PageIndex{4}\): The four bond stretching vibrations in [PtCl4]2- and their irreducible representations. (CC BY-NC-SA; Catherine McCusker)
Figure \(\PageIndex{4}\): The four bond stretching vibrations in [PtCl4]2- and their irreducible representations. (CC BY-NC-SA; Catherine McCusker)By inspection we can see that the first two vibrations (a1g and b2g) won't change the dipole moment of the molecule, and are therefore not infrared active. This can be confirmed with the D4h character table below. The mathematical functions x, y, and z (which represent a change in dipole moment) do not have a1g or b2g symmetry. The functions x and y are degenerate and have eu symmetry. This means the last pair of vibrations will be infrared active, although because they are degenerate they will be at the same energy and appear as a single peak in the IR spectrum.
Raman active vibrations change the polarizability of the molecule, which is more difficult to determine by inspection. Any vibration that changes the polarizability of the molecule will have the same symmetry as the product math functions in the last column of the character table. By consulting the D4h character table we cans see the math functions \(x^2+y^2\) and \(x^2\) have a1g symmetry, the math function \(xy\) has b2g symmetry, and no product functions have eu symmetry. This means that only the first two vibrations (a1g and b2g) will be Raman active and will appear in the Raman spectrum.
You may also note that [PtCl4]2- is a centrosymmetric molecule (has an inversion center) and the two infrared active vibrations belonged to an odd (u) symmetry irreducible representation and the two Raman active vibrations belonged to even (g) symmetry irreducible representations
| D4h | E | 2C4 | C2 | 2C2' | 2C2'' | i | 2S4 | σh | 2σv | 2σd | ||
| A1g | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | \(x^2+y^2\), \(z^2\) | |
| A2g | 1 | 1 | 1 | -1 | -1 | 1 | 1 | 1 | -1 | -1 | Rz | |
| B1g | 1 | -1 | 1 | 1 | -1 | 1 | -1 | 1 | 1 | -1 | \(x^2-y^2\) | |
| B2g | 1 | -1 | 1 | -1 | 1 | 1 | -1 | 1 | -1 | 1 | \(xy\) | |
| Eg | 2 | 0 | -2 | 0 | 0 | 2 | 0 | -2 | 0 | 0 | (Rx, Ry) | (\(xz,yz\)) |
| A1u | 1 | 1 | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 | ||
| A2u | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 | 1 | 1 | \(z\) | |
| B1u | 1 | -1 | 1 | 1 | -1 | -1 | 1 | -1 | -1 | 1 | ||
| B2u | 1 | -1 | 1 | -1 | 1 | -1 | 1 | -1 | 1 | -1 | ||
| Eu | 2 | 0 | -2 | 0 | 0 | -2 | 0 | 2 | 0 | 0 | (\(x,y\)) |
Chirality
Around the year 1847, the French scientist Louis Pasteur provided an explanation for the optical activity of tartaric acid salts. when he carried out a particular reaction, Pasteur observed that two types of crystals precipitated. Patiently and carefully using tweezers, Pasteur was able to separate the two types of crystals. Pasteur noticed that the types rotated the plane polarized by the same amount but in different directions. These two compounds are called enantiomers.
What are Enantiomers?
Two compounds are enantiomers if they are non-superimposable mirror images of each other. As was mentioned, enantiomers are characterized by their ability to rotate plane-polarized light. They also have the same physical properties (e.g., melting point, etc.) relative to each other. As a result, they are also referred to as being optically active. When it comes to symmetry, there are some general rules of thumb that help determine whether a molecule is chiral or achiral. This can be very useful because sometimes molecules can have relatively complicated structures and geometries that knowing whether or not they are chiral becomes a daunting task. The goal, as a result, is to determine the point group of the molecule and the symmetry elements associated with it, then inferring the chirality of the molecule.
Using Symmetry to Determine Chirality
For a molecule to be chiral, it must lack:
- Center of inversion \(i\) and a plane of symmetry \(\sigma\).
- An improper rotation axis (rotation-reflection axis) \(S_{n}\).
However, since, by definition, an improper rotation axis is a rotation about an certain axis followed by reflection about a plane perpendicular to that axis, and an inversion center is simply \(S_{2}\), the absence of an improper axis requires, in most cases, that absence of both a plane of symmetry and an inversion center. As a result, it suffices, in most cases, to check for improper rotation axes to determine whether a molecule is chiral or not.
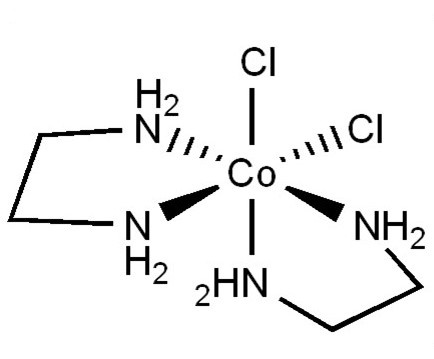
As a result of the previous discussion, there are a few classes of point groups that lack an improper axis. Those classes are C1, Cn, and Dn. cis-dichloridobis(ethylenediamine)cobalt(III) is in the C2 point group and has two enantiomers that are chiral (Figure \(\PageIndex{5}\)). The trans-dichloridobis(ethylenediamine)cobalt(III) is achiral.