
6.2.3: Metal Hydrides
- Page ID
- 360905
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Learning Objectives
In this lecture you will learn the following
- Know about metal hydrides, their synthesis, characterization and their reactivity.
- Know about σ−complexes and their properties.
- Know about transition metal dihydrogen complexes.
Metal hydrides occupy an important place in transition metal organometallic chemistry as the M−H bonds can undergo insertion reactions with a variety of unsaturated organic substrates yielding numerous organometallic compounds with M−C bonds. Not only the metal hydrides are needed as synthetic reagents for preparing the transition metal organometallic compounds but they also are required for important hydride insertion steps in many catalytic processes. The first transition metal hydride compound was reported by W. Heiber in 1931 when he synthesized Fe(CO)4H2. Though he claimed that the Fe(CO)4H2 contained Fe−H bond, it was not accepted until 1950s, when the concept of normal covalent M−H bond was widely recognized.
The metal hydride moieties are easily detectable in 1H NMR as they appear high field of TMS in the region between 0 to 60 ppm, where no other resonances appear. The hydride moieties usually couple with metal centers possessing nuclear spins. Similarly, the hydride moieties also couple with the adjacent metal bound phosphine ligands, if at all present in the complex, exhibiting characteristic cis (J = 15 − 30 Hz) and trans (J = 90 − 150 Hz) coupling constants. In the IR spectroscopy, the M−H frequencies appear between (1500 − 2200) cm−1 but their intensities are mostly weak. Crystallographic detection of metal hydride moiety is difficult as hydrogen atoms in general are poor scatterer of X−rays. Located adjacent to a metal atom in a M−H bond, the detection of hydrogen atom thus becomes challenging and as a consequence the X−ray crystallographic method systematically underestimates the M−H internuclear distance by ~ 0.1 Å. However, better data could be obtained by performing the X−ray diffraction studies at a low temperature in which the thermal motion of the atoms are significantly reduced. In light of these facts, the neutron diffraction becomes a powerful method for detection of the metal hydride moieties as hydrogen scatters neutrons more effectively and hence the M−H bond distances can be measured more accurately. A limitation of neutron diffraction method is that large sized crystals are required for the study.
Synthesis
Following reactions are employed for synthesizing metal hydrides.
i. Protonation reactions
For this reaction to occur the metal center has to be basic and electron rich.
\[\ce{[Fe(CO)4]^{-2} ->[H+] [HFe(CO)4]^{-} ->[H+] H2Fe(CO)4} \nonumber \]
ii. From hydride donors
Generally for this method, a main group hydride is reacted with metal halide.
\[\ce{WCl6 + LiBEt3H + PR3 -> WH6(PR3)3} \nonumber \]
iii. Using dihydrogen (H2) addition
This method involves oxidative addition of H2 and thus requires metal centers that are capable of undergoing the oxidative addition step.
\[\ce{WMe6 + PMe2Ph ->[H_{2}] WH6(PMe2Ph)3} \nonumber \]
iv. From a ligand
This method takes into account the β−elimination that occur in a variety of metal bound ligand moieties, thereby yielding a M−H bond.
\[\ce{RuCl2(PPh3)3 + KOCHMe2 + PPH3 -> RuH2(PPh3)4 + Me2CO + KCl} \nonumber \]
Reactions of metal hydrides
Metal hydrides are reactive species kinetically and thus participate in a variety of transformations like the ones discussed below.
i. Deprotonation reactions
The deprotonation reaction can be achieved by a hydride moiety resulting in the formation of H2gas as shown below.
\[\ce{WH6(PMe3)3 + NaH -> Na[WH5(PMe3)3] + H2} \nonumber \]
ii. Hydride transfer and insertion
In this reaction a hydride transfer from a metal center to formaldehyde resulting in the formation of a metal bound methoxy moiety is observed as shown below.
\[\ce{Cp2*ZrH2 + CH2O -> Cp2*Zr(OMe)2} \nonumber \]
iii. Hydrogen atom transfer reaction
An example of hydrogen atom transfer reaction is given below.
\[\ce{[Co(CN)5H]^{3-} + PhCH=CHCOOH -> [Co(CN)5]^{3-} + PHCH-CH2COOH} \nonumber \]
It is interesting to note that the nature of hydrogen atom in a M−H bond can vary from being protic in nature, when bound to electron deficient metal centers as in metal carbonyl compounds, to that of being hydridic in nature, when bound to more electropositive early transition metals. In the latter case, the hydride moieties tend to be basic and exhibit hydride transfer reactions with electrophiles like aldehydes or ketones. Furthermore, the protonation of these basic metal hydrides leads to the elimination of dihydrogen (H2) gas along with the generation of a vacant coordination site at the metal center.
Bridging hydrides
The metal hydrides usually show two modes of binding, namely terminal and bridging. In case of the bridging hydrides, the hydrogen atom can bridge between two or even more metal centers and thus, the bridging hydrides often display bent geometries.
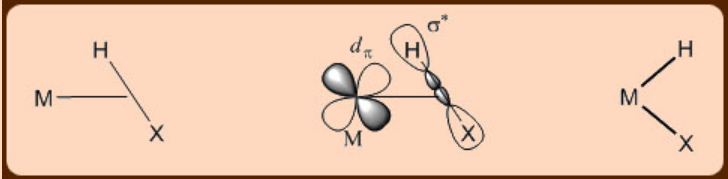
σ−complexes
σ−complexes are rare compounds, in which the σ bonding electrons of a X−H bond further participate in bonding with a metal center (X = H, Si, Sn, B, and P). The σ complexes thus exhibit an askewed binding to a metal center with the hydrogen atom, containing no lone pair, being more close to the metal center and thereby resulting in a side−on structure. Many times if the metal center is electron rich, then further back donation to the σ* orbital of the metal bound X−H moiety may occur resulting in a complete cleavage of the X−H bond.
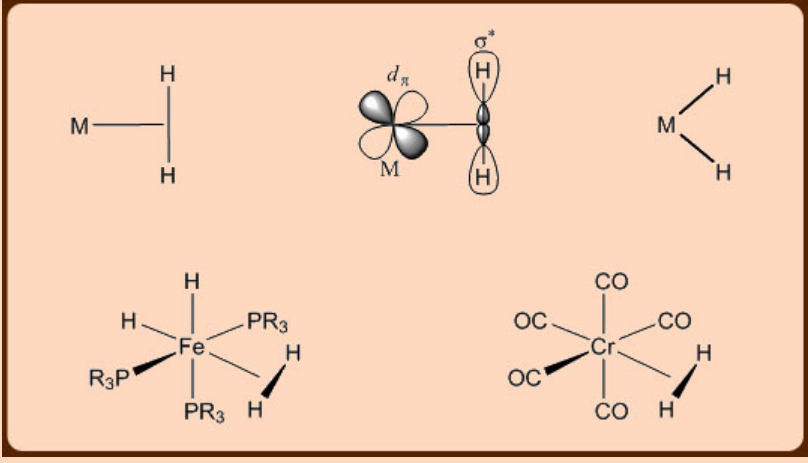
Metal dihydrogen complexes
The simplest variant of a σ−complex contains a dihydrogen ligand. The first dihydrogen complex was isolated by Kubas, after which many new ones were reported.
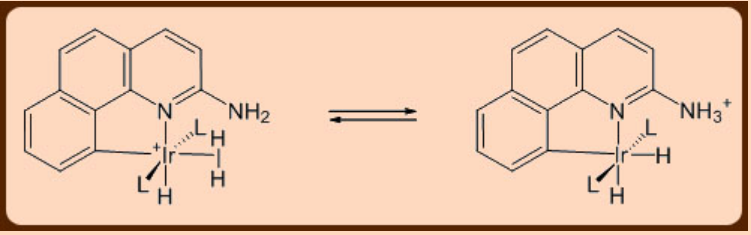
Quite expectedly, the dihydrogen moiety bound to a metal in a σ−complex is found to be more acidic (pKa = 0 − 20) when compared to the free dihydrogen molecule (pKa = 35). It is interesting to note that the pKa change associated with the binding of dihydrogen to a metal in a σ−complex relative to that of the free H2 molecule is significantly larger than the change associated with binding of H2O to metal. Owing to this inherent acidity, the deprotonation of the metal bound dihydrogen moiety by a base can thus be appropriately employed for heterolytic activation of the dihydrogen moiety as illustrated below.
The dihydrogen complexes of metals are often referred to as nonclassical hydrides. The electron rich π basic metals are anticipated to split the metal bound dihydrogen moieties resulting in classical dihydride complexes. Along the same line of thinking, the electron deficient and less π basic metal would tend to stabilize a dihydrogen complex. The dihydrogen complexes can also be characterized by the X ray diffraction as well as neutron diffraction methods. In IR spectrum, the metal bound H−H stretch appear in the range (2300 − 2900) cm−1 while in the 1H NMR spectrum the same appear between 0 to −10 ppm as a broad peak. The dihydrogen complexes are often characterized by isotopic labeling studies of metal bound H−D moiety that shows a coupling constant of 20 – 34 Hz as supposed to 43 Hz observed in case of the free H−D molecule.
Problems
1. Predict the product of the reaction.
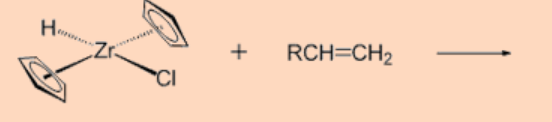
Ans:
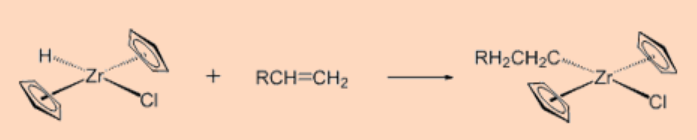
2. Give the oxidation state and total valence electron count of the metal center.
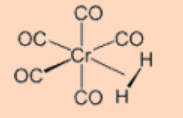
Ans: Oxidation state 0 and 18 VE
3. What kind of metal centers would stabilize metal dihydrogen complexes?
Ans: Electron deficient and less π basic ligands
4. Specify whether the nature of hydrogen moiety in the complex, HCo(CO)4 is acidic or basic?
Ans: Acidic
5. Where do the M−H stretching bands appear in the IR spectrum of metal hydride complexes?
Ans: 1500 to 2200 cm-1
Self Assessment test
1. Predict the product of the reaction.
\[\ce{IrCl(Co)(Pph3)2 ->[H_{2}]} \nonumber \]
Ans:
\[\ce{IrCl(CO)(PPh3)2 ->[H2] IrH2Cl(CO)(PPh3)2} \nonumber \]
2. Give the oxidation state and total valence electron count of the metal center.
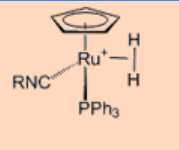
Ans: Oxidation state +2 and 18 VE
3. What kind of metal centers would stabilize classical dihydride complexes?
Ans: Electron rich and more p basic ligands
4. Specify whether the nature of hydrogen moiety in the complex, IrH5(PCy3)2 is acidic or basic?
Ans: Basic
5. Between X−ray diffraction and neutron diffraction, which is a better method for the characterization of the M−H moiety?
Ans: Neutron diffraction
Summary
Metal hydrides are important compounds in the overall scheme of organometallic chemistry as they are involved in many crucial steps of numerous catalytic reactions. Apart from metal hydrides another important class of compounds are transition metal σ−complexes whose simplest variant are the metal dihydrogen complexes. These σ−complexes and the metal dihydrogen complexes are important for the heterolytic activations of the respective metal bound H−heteroatom and the H−H bonds.