
Section 12.6: Electron Transfer
- Page ID
- 440909
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Overview
Metalloproteins containing a single type of redox cofactor can be divided into two general classes: electron carriers and proteins involved in the transport or activation of small molecules. Some of the factors that seem to be characteristic of electron-transfer proteins (these proteins are sometimes called "electron transferases") are: (a) possession of a suitable cofactor to act as an electron sink (metal center with multiple stable oxidation states); (b) placement of the cofactor close enough to the protein surface to allow electrons to move in and out; (c) existence of a hydrophobic shell adjacent to, but not always entirely surrounding, the cofactor; (d) small structural changes accompanying electron transfer; and (e) an architecture that permits slight expansion or contraction in preferred directions upon electron transfer.
Metalloproteins that function as electron transferases typically place their metal coordination sites in a hydrophobic environment and may provide hydrogen bonds (in addition to ligands) to assist in stabilizing both the oxidized and the reduced forms of the cofactor. Metal-ligand bonds remain intact upon electron transfer to minimize inner-sphere reorganization energy. Many of the complex multisite metalloenzymes (e.g., cytochrome c oxidase, xanthine oxidase, the nitrogenase FeMo protein) contain redox centers that function as intramolecular electron transferases, shuttling electrons to/from other metal centers that bind exogenous ligands during enzymatic turnover.
There are three classes of metal containing electron transferases, each of which contains many members that exhibit important structural differences: blue copper proteins, iron-sulfur proteins, and cytochromes.
Blue Copper Proteins
Blue copper proteins were first isolated from bacteria in the 1950s and from plant tissues in the early 1960s. The intense blue color of these proteins is due to a strong absorption band at a wavelength of about 600 nm. Although simple Cu2+ complexes, such as [Cu(H2O)6]2+ and [Cu(NH3)4]2+, are also blue due to an absorption band at 600 nm, the intensity of the absorption band is about 100 times less than that of a blue copper protein. Moreover, the reduction potential for the Cu2+/Cu+ couple in a blue copper protein is usually +0.3 to +0.5 V, considerably more positive than that of the aqueous Cu2+/Cu+ couple (+0.15 V).
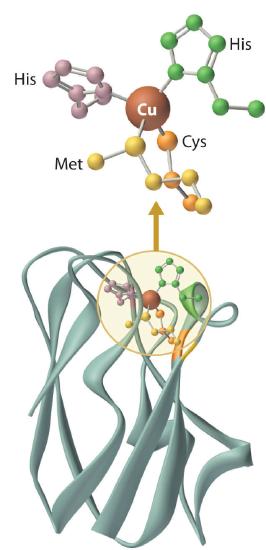
The copper center in blue copper proteins has a distorted tetrahedral structure, in which the copper is bound to four amino acid side chains (Figure \(\PageIndex{3}\)). Although the most common structures for four-coordinate Cu2+ and Cu+ complexes are square planar and tetrahedral, respectively, the structures of the oxidized (Cu2+) and reduced (Cu+) forms of the protein are essentially identical. Thus the protein forces the Cu2+ ion to adopt a higher-energy structure that is more suitable for Cu+, which makes the Cu2+ form easier to reduce and raises its reduction potential.
Moreover, by forcing the oxidized and reduced forms of the metal complex to have essentially the same structure, the protein ensures that electron transfer to and from the copper site is rapid because only minimal structural reorganization of the metal center is required. Kinetics studies on simple metal complexes have shown that electron-transfer reactions tend to be slow when the structures of the oxidized and reduced forms of a metal complex are very different, and fast when they are similar. You will see that other metal centers used for biological electron-transfer reactions are also set up for minimal structural reorganization after electron transfer, which ensures the rapid transfer of electrons.
The blue copper proteins are characterized by intense S(Cys) → Cu chargetransfer absorption near 600 nm, an axial EPR spectrum displaying an unusually small hyperfine coupling constant, and a relatively high reduction potential.4,8-10 With few exceptions (e.g., photosynthetic organisms), their precise roles in bacterial and plant physiology remain obscure. X-ray structures of several blue copper proteins indicate that the geometry of the copper site is approximately trigonal planar, as illustrated by the Alcaligenes denitrificans azurin structure (Figure 6.5).11,12 In all these proteins, three ligands (one Cys, two His) bind tightly to the copper in a trigonal arrangement. Differences in interactions between the copper center and the axially disposed ligands may significantly contribute to variations in reduction potential that are observed12 for the blue copper electron transferases. For example, E°' = 276 mV for A. denitrificans azurin, whereas that of P. vulgaris plastocyanin is 360 mV. In A. denitrificans azurin, the Cu-S(Met) bond is 0.2 Å longer than in poplar plastocyanin, and there is a carbonyl oxygen 3.1 Å from the copper center, compared with 3.8 Å in plastocyanin. These differences in bond lengths are expected to stabilize Cull in azurin to a greater extent than in plastocyanin, and result in a lower E°' value for azurin.
Iron Sulfur Proteins
Although all known bacteria, plants, and animals use iron–sulfur proteins to transfer electrons, the existence of these proteins was not recognized until the late 1950s. Iron–sulfur proteins transfer electrons over a wide range of reduction potentials, and their iron content can range from 1 to more than 12 Fe atoms per protein molecule. In addition, most iron–sulfur proteins contain stoichiometric amounts of sulfide (S2−).
These properties are due to the presence of four different kinds of iron–sulfur units, which contain one, two, three, or four iron atoms per Fe–S complex (Figure \(\PageIndex{5}\)). In all cases, the Fe2+ and Fe3+ ions are coordinated to four sulfur ligands in a tetrahedral environment. Due to tetrahedral coordination by weak-field sulfur ligands, the iron is high spin in both the Fe3+ and Fe2+ oxidation states, which results in similar structures for the oxidized and reduced forms of the Fe–S complexes. Consequently, only small structural changes occur after oxidation or reduction of the Fe–S center, which results in rapid electron transfer.
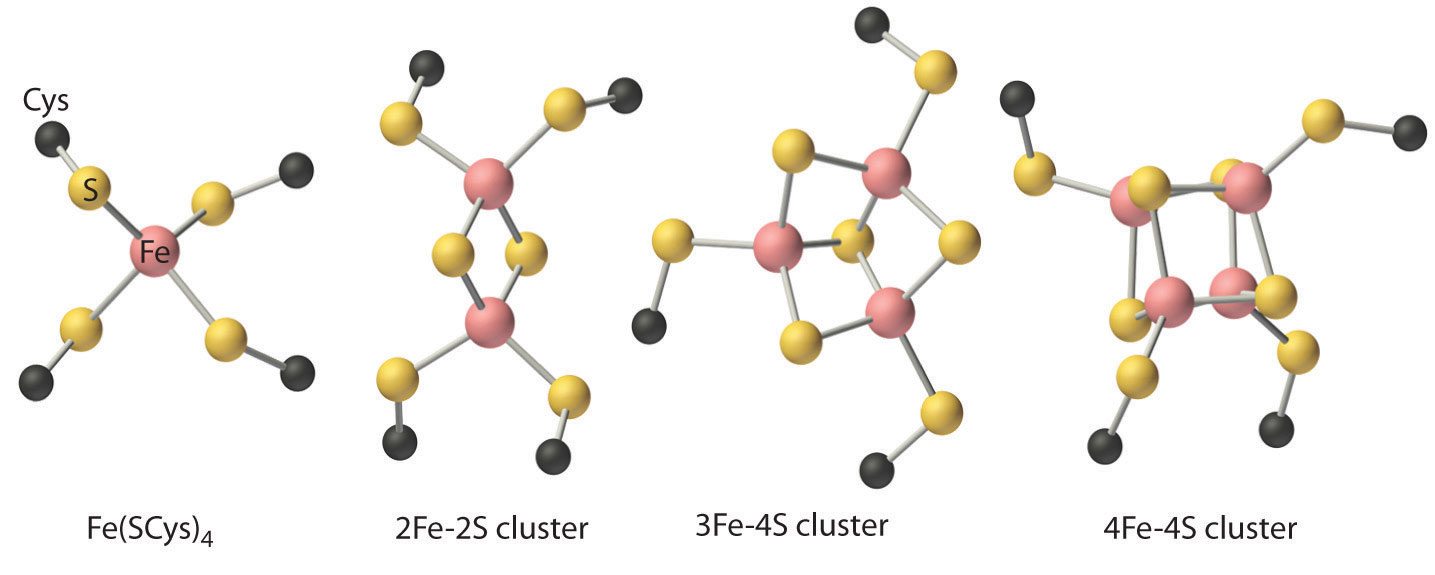
The iron-sulfur proteins play important roles13,14 as electron carriers in virtually all living organisms, and participate in plant photosynthesis, nitrogen fixation, steroid metabolism, and oxidative phosphorylation, as well as many other processes (Chapter 7). The optical spectra of all iron-sulfur proteins are very broad and almost featureless, due to numerous overlapping charge-transfer transitions that impart red-brown-black colors to these proteins. On the other hand, the EPR spectra of iron-sulfur clusters are quite distinctive, and they are of great value in the study of the redox chemistry of these proteins.
The simplest iron-sulfur proteins, known as rubredoxins, are primarily found in anaerobic bacteria, where their function is unknown. Rubredoxins are small proteins (6 kDa) and contain iron ligated to four Cys sulfurs in a distorted tetrahedral arrangement. The E°' value for the FeIII/II couple in water is 770 mV; that of C. pasteurianum rubredoxin is -57 mV. The reduction potentials of iron-sulfur proteins are typically quite negative, indicating a stabilization of the oxidized form of the redox couple as a result of negatively charged sulfur ligands.
The [2Fe-2S] ferredoxins (10-20 kDa) are found in plant chloroplasts and mammalian tissue. The structure of Spirulina platensis ferredoxin15 confirmed earlier suggestions, based on EPR and Mössbauer studies, that the iron atoms are present in a spin-coupled [2Fe-2S] cluster structure. One-electron reduction (E°' ~ -420 mV) of the protein results in a mixed-valence dimer (Equation 6.3):
\[[Fe_{2}S_{2}(SR)_{4}]^{2-} \xrightleftharpoons[-e^{-}]{+e^{-}} [Fe_{2}S_{2}(SR)_{4}]^{3-} \tag{6.3}\]
\[Fd_{ox} \qquad \qquad \qquad Fd_{red}\]
\[2Fe(III) \qquad \qquad Fe(II) + Fe(III)\]
The additional electron in Fdred is associated with only one of the iron sites, resulting in a so-called trapped-valence structure.16 The [Fe2S2(SR)4]4- cluster oxidation state, containing two ferrous ions, can be produced in vitro when strong reductants are used.
Four-iron clusters [4Fe-4S] are found in many strains of bacteria. In most of these bacterial iron-sulfur proteins, also termed ferredoxins, two such clusters are present in the protein. These proteins have reduction potentials in the -400 mV range and are rather small (6-10 kDa). Each of the clusters contains four iron centers and four sulfides at alternate comers of a distorted cube. Each iron is coordinated to three sulfides and one cysteine thiolate. The irons are strongly exchange-coupled, and the [4Fe-4S] cluster in bacterial ferredoxins is paramagnetic when reduced by one electron. The so-called "high-potential ironsulfur proteins" (HiPIPs) are found in photosynthetic bacteria, and exhibit anomalously high (~350 mV) reduction potentials. The C. vinosum HiPIP (10 kDa) structure demonstrates that HiPIPs are distinct from the [4Fe-4S] ferredoxins, and that the reduced HiPIP cluster structure is significantly distorted, as is also observed for the structure of the oxidized P. aerogenes ferredoxin. In addition, oxidized HiPIP is paramagnetic, whereas the reduced protein is EPR-silent.
This bewildering set of experimental observations can be rationalized in terms of a "three-state" hypothesis (i.e., [4Fe-4S(SR)4]n- clusters exist in three physiological oxidation states).17 This hypothesis nicely explains the differences in magnetic behavior and redox properties observed for these iron-sulfur proteins (Equation 6.4):
\[[4Fe-4S(SR)_{4}]^{-} \xrightleftharpoons[-e^{-}]{+e^{-}} [4Fe-4S(SR)_{4}]^{2-} \xrightleftharpoons[-e^{-}]{+e^{-}} [4Fe-4S(SR)_{4}]^{3-} \tag{6.4}\]
\[HiPIP_{ox} \qquad \qquad \qquad HiPIP_{red} \qquad \qquad \qquad Ferredoxin_{red}\]
\[Ferredoxing_{ox}\]
The bacterial ferredoxins and HiPIPs all possess tetracubane clusters containing thiolate ligands, yet the former utilize the -2/-3 cluster redox couple, whereas the latter utilize the -1/-2 cluster redox couple.
The protein environment thus exerts a powerful influence over the cluster reduction potentials. This observation applies to all classes of electron transferases—the factors that are critical determinants of cofactor reduction potentials are poorly understood at present but are thought18 to include the low dielectric constants of protein interiors (~4 for proteins vs. ~78 for H2O), electrostatic effects due to nearby charged amino-acid residues, hydrogen bonding, and geometric constraints imposed by the protein.
Cytochromes
The cytochromes (from the Greek cytos, meaning “cell”, and chroma, meaning “color”) were first identified in the 1920s by spectroscopic studies of cell extracts. Based on the wavelength of the maximum absorption in the visible spectrum, they were classified as cytochromes a (with the longest wavelength), cytochromes b (intermediate wavelength), and cytochromes c (shortest wavelength). It quickly became apparent that there was a correlation between their spectroscopic properties and other physical properties. For examples, cytochromes c are generally small, soluble proteins with a reduction potential of about +0.25 V, whereas cytochromes b are larger, less-soluble proteins with reduction potentials of about 0 V.
All cytochromes contain iron, and the iron atom in all cytochromes is coordinated by a planar array of four nitrogen atoms provided by a cyclic tetradentate ligand called a porphyrin. The iron–porphyrin unit is called a heme group. The structures of a typical porphyrin (protoporphyrin IX) and its iron complex (protoheme) are shown here. In addition to the four nitrogen atoms of the porphyrin, the iron in a cytochrome is usually bonded to two additional ligands provided by the protein, as shown in Figure \(\PageIndex{4}\).
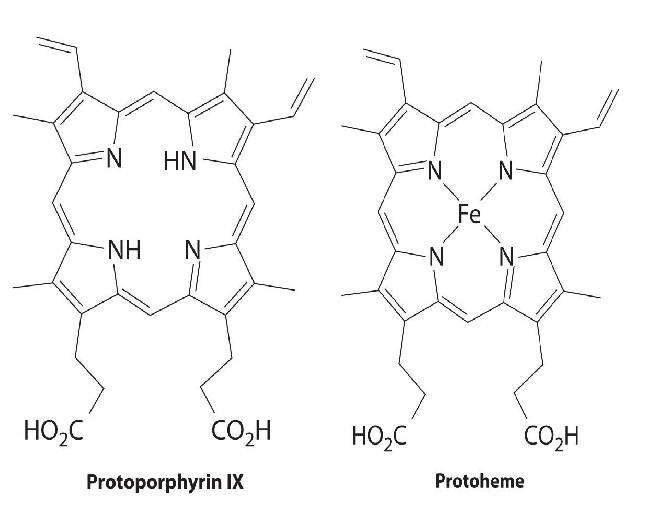
A cytochrome. Shown here is protoporphyrin IX and its iron complex, protoheme.
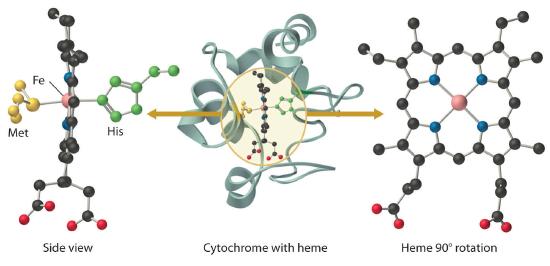
In contrast to the blue copper proteins, two electron configurations are possible for both the oxidized and reduced forms of a cytochrome, and this has significant structural consequences. Thus Fe2+ is d6 and can be either high spin (with four unpaired electrons) or low spin (with no unpaired electrons). Similarly, Fe3+ is d5 and can also be high spin (with five unpaired electrons) or low spin (with one unpaired electron). In low-spin heme complexes, both the Fe2+ and the Fe3+ ions are small enough to fit into the “hole” in the center of the porphyrin; hence the iron atom lies almost exactly in the plane of the four porphyrin nitrogen atoms in both cases. Because cytochromes b and c are low spin in both their oxidized and reduced forms, the structures of the oxidized and reduced cytochromes are essentially identical. Hence minimal structural changes occur after oxidation or reduction, which makes electron transfer to or from the heme very rapid.
Electron transfer reactions occur most rapidly when minimal structural changes occur during oxidation or reduction
As a class, the cytochromes19-22 are the most thoroughly characterized of the electron transferases. By definition, a cytochrome contains one or more heme cofactors. These proteins were among the first to be identified in cellular extracts because of their distinctive optical properties, particularly an intense absorption in the 410-430 nm region (called the Soret band). Cytochromes are typically classified on the basis of heme type. Figure 6.6 displays the three most commonly encountered types of heme: heme a possesses a long phytyl "tail" and is found in cytochrome c oxidase; heme b is found in b-type cytochromes and globins; heme c is covalently bound to c-type cytochromes via two thioether linkages. Cytochrome nomenclature presents a real challenge! Some cytochromes are designated according to the historical order of discovery, e.g., cytochrome c2 in bacterial photosynthesis. Others are designated according to the \(\lambda_{max}\)of the \(\alpha\) band in the absorption spectrum of the reduced protein (e.g., cytochrome c551).
Cytochromes c are widespread in nature. Ambler23 divided these electron carriers into three classes on structural grounds. The Class I cytochromes c contain axial His and Met ligands, with the heme located near the N-terrninus of the protein. These proteins are globular, as indicated by the ribbon drawing of tuna cytochrome c (Figure 6.7). X-ray structures of Class I cytochromes c from a variety of eukaryotes and prokaryotes clearly show an evolutionarily conserved "cytochrome fold," with the edge of the heme solvent-exposed. The reduction potentials of these cytochromes are quite positive (200 to 320 mV). Mammalian cytochrome c, because of its distinctive role in the mitochondrial electron-transfer chain, will be discussed later.
Class II cytochromes c (E°' ~ -100 mV) are found in photosynthetic bacteria, where they serve an unknown function. Unlike their Class I cousins, these c-type cytochromes are high-spin: the iron is five-coordinate, with an axial His ligand. These proteins, generally referred to as cytochromes c' , are four-\(\alpha\)-helix bundles (Figure 6.8). The vacant axial coordination site is buried in the protein interior.
Finally, Class III cytochromes c, also called cytochromes c3, contain four hemes, each ligated by two axial histidines. These proteins are found in a restricted class of sulfate-reducing bacteria and may be associated with the cytoplasmic membrane. The low molecular weights of cytochromes c3 (~14. 7 kDa) require that the four hemes be much more exposed to the solvent than the hemes of other cytochromes (see Figure 6.9), which may be in part responsible for their unusually negative (-200 to -350 mV) reduction potentials. These proteins possess many aromatic residues and short heme-heme distances, two properties that could be responsible for their anomalously large solid-state electrical conductivity.24