
7.6: FDA Drug Approval Process
- Page ID
- 227700

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)
Before an investigational new drug (IND) can even be tested on humans, extensive pre-clinical research must be conducted. This includes first identifying potential drug candidates:
- Sometimes a drug is discovered purely by serendipity,
- Other times natural products produced by microorganisms are screened for antifungal, antibiotic, antiviral, or antitumor activity.
- Another approach is to use computer modeling to design drugs that will interact effectively with a particular receptor in the body—for example, DNA or a specific enzyme.
- Once an active compound or a target has been identified, many new compounds—hundreds, even thousands—are synthesized. These hundreds or thousands of compounds are then usually screened in vitro (generally, using cell cultures in a Petri dish—that is, outside the living organism). The most active compounds are then screened in vivo (using animal assays). The drug candidate is tested on at least two different species of animals (one rodent and one non-rodent) because drugs do not always affect different species the same way. (For example, cisplatin was tested on both mice and dogs.) Both short- and long-term testing is conducted on animals. Short-term testing lasts from 2 weeks to 3 months and is designed to examine the metabolism and toxicity of the drug candidate. Long-term testing lasts from a few weeks to several years and is done to see whether long-term use of the drug will lead to cancer or birth defects.
After the completion of pre-clinical research, the FDA meets with the sponsor of the drug (usually a pharmaceutical company—in the case of cisplatin, Bristol-Myers), and the sponsor submits data demonstrating that the drug candidate is both biologically active and safe for administration to humans. After these meetings, the sponsor submits the drug candidate as an IND, and clinical testing can begin. The purpose of clinical research is to determine the safety and efficacy of the IND for the treatment of a particular disease or condition in humans. Clinical research is divided into three phases in the normal course of testing. (Certain drugs are placed in an accelerated development and review process; this will not be discussed here, but is described more fully on the FDA website on the new drug development process
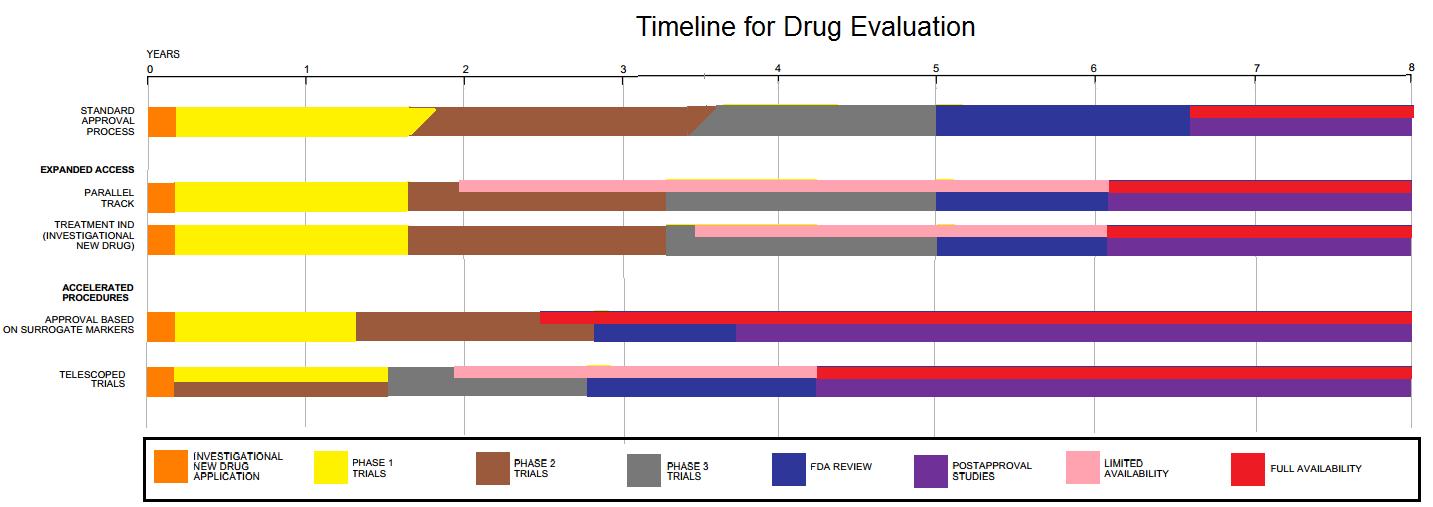
- Phase 1 clinical studies represent the first time that an IND is tested on humans—generally, healthy volunteers, but sometimes patients (the latter was the case with phase 1 clinical studies of cisplatin). The purpose of these studies is to determine the metabolism, structure-reactivity relationships, mechanism of action, and side effects of the drug in humans. If possible, phase 1 studies are used to ascertain the efficacy of the drug. Phase 1 studies are usually conducted on 20 to 80 subjects.
- The purpose of phase 2 clinical trials is to determine the efficacy of a drug to treat patients with a specific disease or condition, as well as common short-term side effects or risks. These studies are conducted on a larger scale than phase 1 studies and typically involve several hundred patients.
- Phase 3 clinical trials provide more information about the efficacy and safety of the drug and allow scientists to extrapolate the results of clinical studies to the general population. Phase 3 studies generally involve several hundred to several thousand people.
There are several checks and balances in the process of clinical trials; among them is the use of institutional review boards (IRBs) and advisory committees. IRBs are designed to protect the rights and welfare of people participating in clinical trials both before and during the trials. IRBs comprise at least five experts and lay people with a variety of backgrounds to provide a complete review of clinical proceedings. In addition, the CDER uses advisory committees comprising various experts in order to obtain outside opinions and advice about a new drug, a new indication for a previously approved drug, labeling information about a drug, guidelines for developing particular kinds of drugs, or the adequacy of safety and efficacy data. The sponsor of a drug makes a formal application to the FDA to approve a new drug for use in the United States by submitting a new drug application (NDA). An NDA must include results and analyses from tests of the drug on both animals and humans, as well as a description of how the drug was manufactured. The NDA must provide enough information for FDA reviewers to make several critical decisions, including whether the drug is safe and efficacious and whether its benefits outweigh its risks, whether the drug's labeling information is appropriate, and whether the manufacturing methods used to obtain the drug are adequate for ensuring the purity and integrity of the drug. The process of developing and testing a new drug is a lengthy one. The FDA estimates that it takes a little over 8 years to test a drug—including early laboratory and animal testing—before the final approval for use by the general public.
Inside the FDA—Vocabulary CDER: Center for Drug Evaluation and Research FDA: Food and Drug Administration IND: Investigational New Drug IRB: Institutional Review Board NDA: New Drug Application
Sources
Drug Development. Wikipedia Commons. Under under the Creative Commons Attribution-ShareAlike License.
Image by Kernsters - Graph created based on information provided in Scientific American article, "Faster Evaluation of Vital Drugs". Fro Wikipedia Commons under CC BY-SA 3.0 license.