
Lab 1: Cyclic Voltammetry
- Page ID
- 204117
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)GOALS
- Students should be able to optimize the experimental condition to produce the best waveform.
- Students should be able to extract key information from a CV waveform. (i.e. peak current and peak potential)
- Students should be able to use the extracted information for practical application. (i.e. determining the unknown concentration of a compound)
Introduction
Cyclic voltammetry (CV) is a technique used to study reaction mechanisms that involve the transferring of electrons. The method involves linearly varying an electrode potential between two limits at a specific rate while monitoring the current that develops in an electrochemical cell. This experiment is performed under conditions where voltage is in excess of that predicted by the Nernst equation (Equation \(\ref{1.1}\)).¹ Although CV is best at providing qualitative information about reaction mechanisms, several quantitative properties of the charge transfer reaction can also be determined.
\[ E=E° -\dfrac{RT}{nF} \ln Q \label{1.1}\]
Cyclic voltammetry involves applying a voltage to an electrode immersed in an electrolyte solution, and seeing how the system responds. In CV, a linear sweeping voltage is applied to an aqueous solution containing the compound of interest. A linear sweeping voltage is defined by the voltage (or potential) being varied linearly at the speed of the scan rate. The variation of the voltage can be seen in Figure 1.1. The voltage is initially given by Equation \(\ref{1.4}\) (see below). After the voltage reaches a certain maximum value, the potential is reversed and the sign of vt reverses and Ei becomes the maximum voltage, \(E_\lambda \). The switch takes place at the peak which can be seen in Figure 1.1. The process can then be repeated in a periodic, or cyclic manner. The voltage after the potential sweep direction is switched is given by Equation \(\ref{1.5}\).
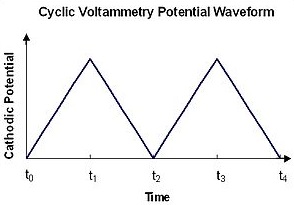
Figure 1.1: Potential versus time program for cyclic voltammetry showing the forward and reversed linear potential ramp.
As an important tool for studying mechanisms and rates of oxidation and reduction processes, CV provides the capability for generating a species during the forward scan and then probing its fate with the reverse scan or subsequent cycles. This process can occur within a few seconds. There is a unique aspect of cyclic voltammetry: three electrodes used. The electrodes are a working electrode, a reference electrode, and a counter electrode. The working electrode can be seen as a medium whose reductive or oxidative power can be externally adjusted by the magnitude of the applied potential. As the potential is increased or decreased linearly versus time, the working electrode becomes a stronger oxidant or reductant, respectively. Therefore, the working electrode, which typically consists of a chemically inert conductive material such as platinum, acts as a donor or acceptor of electrons. The general reaction written in Reaction \(\ref{1.2}\) refers to the addition of electrons to the oxidized electrode and the electrode transforming to its most reduced form.
\[ O + ne^- \rightarrow R \label{1.2}\]
The reference electrode, typically AgCl or calomel, keeps the potential between itself and the working electrode constant. The potential is measured between the reference and working electrodes, and the current is measured between the working and counter electrodes. A counter electrode is employed to allow for accurate measurements to be made between the working and reference electrodes. The counter electrode's role is to ensure that the current does not run through the reference electrode since such a flow would change the reference electrodes potential. A voltage sweep from Ei (initial voltage) to Ef (final voltage) is produced using a signal generator. The voltage is applied to the working electrode using a potentiostat. A potentiostat is an external power source. By sweeping the voltage slowly, information may be extracted from a graph of potential versus current going through the sample. Polarography utilizes this method of analysis where the limited current arising from a redox process in the solution during the sweep. This information is used to quantitatively determine the concentration of species that are electrochemically active in solution.
CV differs from polarography in two important ways. Firstly, the working electrode at which the reactions of interest occur has a constant area, not one which changes with time as in classical polarography. This electrode may be a solid such as graphite or platinum with a small surface area, or a stationary or hanging mercury drop. The latter type of electrode may have its surface renewed periodically. The second difference is that the potential of the working electrode is scanned rapidly over a wide potential range and then returned to its initial value using an applied potential signal which varies linearly with time between the initial value and the final value at the limit of the forward scan. Normally, this technique is applied so that currents due to reduction processes are observed during the forward scan and those due to oxidation on the reverse scan. The resulting cyclic voltammogram from a typical potential against time profile is shown in Figure 1.2.
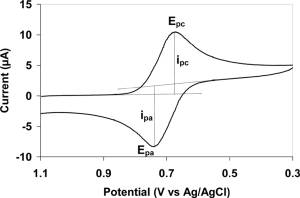
Figure 1.2: Cyclic Voltammogram of measured current versus applied potential. This diagram also shows the points where the y-values of peak anodic current (ipa) and peak cathodic current (ipc) as well as the x-values of peak anodic potential (Epa) and peak cathodic potential (Epc).
Guided Example
Consider the response of the electron transfer reaction
\[ A + ne^- \rightleftharpoons B^- \label{1.3}\]
to the application of an electrode potential which is varying linearly with time. The electrode potential is given by the equation
\[ E = E_i - vt \label{1.4}\]
where Ei is the initial potential, v is the potential sweep rate (in volts s-1) and t is time after the start of voltage sweep. After reaching some time, λ, the direction of the potential sweep is switched, and the equation describing the electrode potential becomes:
\[ E = E_\lambda + v(t - \lambda) \label{1.5}\]
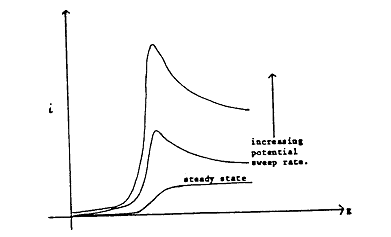
where Eλ is the value of E at the switching point. Considering that the initial sweep is in the negative direction where reduction reactions are expected, it is clear that, if the sweep rate is sufficiently slow, the current against potential curve approaches that obtained by steady-state measurements. However, as v is increased, a peak develops on the i-E curve which becomes increasingly prominent (Figure 1.3). The peak is produced from the combined effects of high mass transfer rates in the non-steady state followed by the progressive depletion of the reactant in the diffusion layer.
{kind=link}
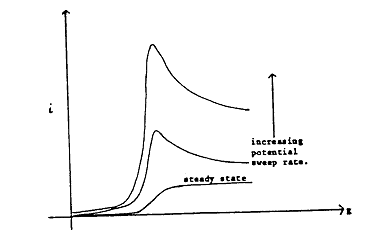
Figure 1.3. Effect of potential sweep rate on the PE curves in a linear potential sweep experiment adjacent to the electrode.
It should be noted that since E is a linear function of t, the potential axis is also a time axis.
In order to relate the observed current to the reactant concentration cA, one must know how cA varies with distance from the electrode, x, and with time, t. This variation is described by Fick's Law for mass transfer by diffusion:
\[ \dfrac{\partial c_A}{\partial t} = D_A \dfrac{\partial^2c_A}{\partial x^2} \label{1.6} \]
where DA is the diffusion coefficient of reactant A (units: cm2/s). The solution of this second order partial differential equation requires specification of boundary and initial conditions and is described in textbooks on electroanalytical chemistry1,2. If the electron transfer reaction is sufficiently fast in Reaction \(\ref{1.3}\) to maintain a Nernstian equilibrium at the electrode surface (i.e., the reversible case), then the peak current, ip for a negative sweep is given by Equation \(\ref{1.7}\). The variables are described in Table 1.1.
\[ i_p = (2.69 \times 10^5) \; n^{3/2} \; SD_A^{1/2} \; v^{1/2} \; C_A, \label{1.7} \]
Table 1.1: Variables of the modified Fick's Law, Equation \(\ref{1.7}\)
| Variable | Description | Units |
|---|---|---|
| \( i_p \) | peak current | A |
| n | number of electrons transferred | - |
| S | Surface Area |
cm2 |
| DA | Diffusion coefficient | cm2/s |
| v | scan rate | V/s |
|
cA |
the concentration of compound A in the bulk solution | mole/cm3 |
You may notice that the peak potential is independent of sweep rate and is related to the half-wave potential (\(E_{1/2} \)) by
\[ E_p = E_{1/2} - \dfrac{0.0285}{n} \label{1.8} \]
Furthermore, the shape of the peak is defined by the potential difference between the peak and the position when the current is one half of that at the peak:
\[ E_p = E_{p/2} - \dfrac{0.0565}{n} \label{1.9} \]
Thus, using the measured values of ip, Ep, and Ep/2, one can determine n and DA for a given electrode area and sweep rate. If the electrode reaction is slow so that the surface concentrations of \(A\) and \(B\) are no longer related by the Nernst equation at a given sweep rate, then the peak characteristics change such that
\[ E_p - E_{p/2} > \dfrac{0.0565}{n} \label{1.10} \]
and the peak potential now depends on the sweep rate. Using the appropriate boundary conditions to describe the rate of Reaction \(\ref{1.3}\) in the forward direction, Equation \(\ref{1.6}\) may be solved to obtain expressions for \(i_p\) and \(E_p\) which are now much more complicated.
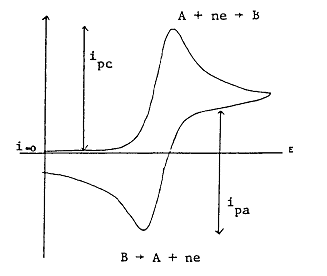
After the direction of the potential sweep is reversed, a second current peak is observed corresponding to oxidation of the product B. When Reaction \(\ref{1.3}\) is reversible, implying that \(B\) is stable, the height of this peak is equal to that observed on reduction, but with the current flowing in the opposite direction. The method of estimating peak currents is illustrated in Figure 1.4, where \(i_{pc}\) is the peak potential in the cathodic sweep.
{kind=link}
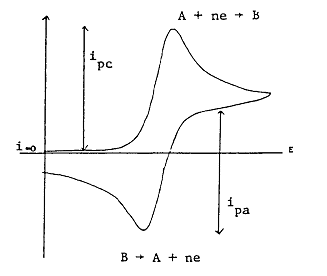
Figure 1.4: CV Waveform of Reaction \(\ref{1.3}\)
The CV waveform in the above figure for the process Reaction \ (\ref{1.3}\) and assumes that only A is initially present in the solution. \(\Delta{E}_p\) is then defined as
\[ \Delta{E}_p = |E_{pc} - E_{pa}|. \label{1.11}\]
When the electrode process is reversible and it independent of sweep speed and is described by Equation \(\ref{1.12}\).
\[ \Delta{E}_p = \dfrac{0.0565}{n} \label{1.12}\]
As v is increased to the stage at which Nernstian equilibrium for Reaction \(\ref{1.3}\) cannot be maintained, ΔEp increases with increasing sweep rate, and the shape and position of the peaks depend on both v and the kinetic parameters of the electrode reaction.
Reaction Mechanisms
One of the major uses of cyclic voltammetry is in the rapid qualitative identification of electrode reaction mechanisms. Organic molecules often undergo a rapid chemical reaction with the solvent or some other constituent of the solution after the electron transfer process. The resulting reaction scheme, referred to as the ECE (electrode-chemical-electrode) mechanism, can be written
\[ A + ne^- \rightarrow B^{n-} \label{1.13}\]
\[ B + Z \rightarrow C \label{1.14}\]
\[ C + me^- \rightarrow D^{m-} \label{1.15}\]
where \(Z\) is the solvent or some other species. The first and third reactions are labeled \(E\) since they involve the electrode, and the second step (or any other chemical step) is labeled \(C\). Hence, the above three-step mechanism is referred to as the ECE reaction mechanism. It is possible to garner information about the (non-electrode-dependent) rate constant for step 2 (Reaction \(\ref{1.14}\)) via cyclic voltammetry.
The standard potential for Reaction \(\ref{1.13}\) is generally different from that for Reaction \(\ref{1.15}\). A typical current-potential curve for such a system is shown in Figure 1.5. The current on the reverse sweep will depend on the sweep rate and the rate constant for Reaction \(\ref{1.14}\, which is assumed to take place under pseudo first-order conditions (\(c_Z \gg c_B\)). For very fast sweep rates, very little \(B\) will react to form \(C\), and the CV waveform will have the same appearance as the reversible case, with reduction and oxidation peaks at I and II, respectively. As \(v\) is decreased, peak II diminishes more rapidly and peak I less rapidly than the usual. The dependence v1/2 would predict this because the chemical step removing species B becomes important and peak I has a contribution from Reaction \(\ref{1.15}\). In addition, a peak develops at III due to oxidation of D.
{kind=link}
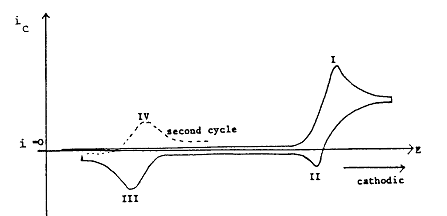
Figure 1.5: Cyclic voltammogram at an intermediate sweep rate for a system with an ECE mechanism.
It should also be noted that the current-potential curves on the second and successive sweeps are not the same as that observed on the first. At very slow sweep rates, peak II disappears completely and peak I then corresponds to the process
\[ A + (m + n)e^- \rightarrow D \label{1.16}\]
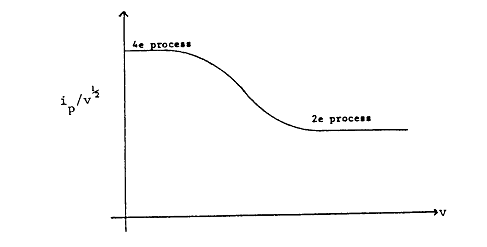
The variation of peak I with sweep rate is shown in Figure 1.6 for the case that \(n = m = 2\).
{kind=link}
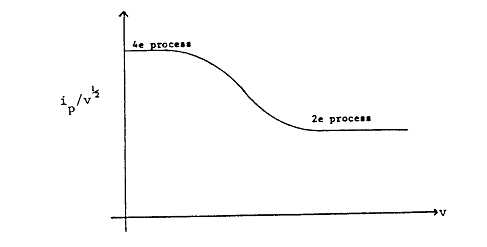
Figure 1.6: Variation of measured current for peak I from Figure 1.4 vs. potential scan rate, \(v\).
The rate constant k for the chemical Reaction \(\ref{1.16}\) can be obtained from an analysis of data obtained in the transition region of Figure 1.6 or from the ratio of peaks I and II at intermediate sweep rates in Figure 1.5.
Cyclic voltammetry can be applied to the analysis of many other reaction mechanisms including those with dimerization of the product of electron transfer, with preceding chemical steps, catalytic processes, etc.
The Instrument
The instrument used for this experiment is the BAS Epsilon potentiostat. It is controlled from a computer running Windows. A number of different electrochemical techniques are available in the Epsilon software, including cyclic voltammetry (current vs. potential for a linear potential sweep), chronoamperometry, time base, bulk electrolysis (current vs. time at a constant potential), and chronopotentiometry (potential vs. time at a fixed current).


Figure 1.7: UCD cyclic voltammetry Instrument (including potentiostat)
To begin an experiment, make sure that the Epsilon unit is turned on, and doubleclick the Epsilon icon on the desktop. Select New from the File menu or click the New icon. This will generate a menu that lists the available techniques. (This list can also be generated by selecting Select NEW Experiment from the Experiment menu or by using the F2 key.)

Figure 1.8: Computer Program
{kind=link}
Highlight Cyclic Voltammetry (CV), and click Select to confirm the selection. An experiment window containing an empty axis set is displayed (Figure 1.9), and the appropriate parameters are set in the various dialog boxes.
Figure 1.9: Computer Program
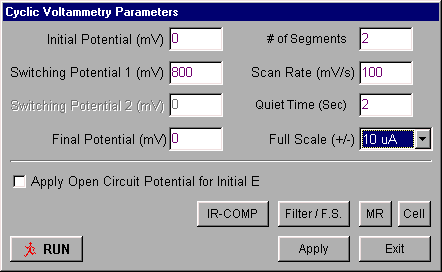
The potential limits and the scan rate for CV are set using the Change Parameters dialog box (Figure 1.10) in either the Experiment menu or the pop-up menu (the pop-up menu is accessed with the right mouse button).
{kind=link}
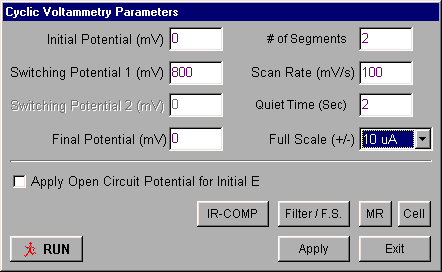
Figure 1.10: Change Parameters dialog box for cyclic voltammetry.
- Potential values are entered in mV, and the Scan Rate in mV/s.
- If the Apply Open Circuit Potential for Initial E box is checked, then the open circuit potential will automatically be measured and used as the Initial Potential.
- When the experiment is started, the cell is held at the Initial Potential for the number of seconds defined by the Quiet Time.
- There are two gain stages for the current-to-voltage converter. The default values of these stages that are used for a given current Full-Scale value are determined by the software. However, they can be adjusted manually using the Filter / F.S. dialog box. This dialog box is also used to change the analog Noise Filter Value settings from the default values set by the software. The Full-Scale value should be at the 10 mA/V setting at the start of the experiment, and then adjusted to a more convenient range depending on the maximum current observed in the experiment. Ask your TA about scale values to better observe your results.
- The default condition of the cell is that the cell is On (i.e., the electronics are connected to the electrodes) during the experiment, and is Off between experiments. THIS OPTION SHOULD NOT BE CHANGED SINCE CONNECTING OR DISCONNECTING THE ELECTRODES WHEN THE CELL IS ON CAN RESULT IN DAMAGE TO THE POTENTIOSTAT, THE CELL, AND/OR THE USER!
- Clicking the IR-COMP button activates the iR compensation option (compensates for the drop in voltage due to the resistance of the solution).
- Clicking Exit will exit the dialog box without saving any changes made to the parameter values. Any changes can be saved by clicking Apply before exiting.
- Range of allowed parameter values:
- Potential = -3275 - +3275 mV
- Scan Rate = 1 - 10,000 mV/s (also see below)
- Quiet Time = 0 - 100 s
- The # of Segments is limited by the total number of data points that can be stored (32,000) (note that in this initial version, the potential resolution of the current measurement is fixed at 1 mV).
- Once the parameters have been set, the experiment can be started by clicking Run (either in this dialog box, in the Experiment menu, in the pop-up menu, on the Tool Bar, or using the F5 key).
Experimental Procedure
Part 1: Reduction of the Ferricyanide Anion
In this portion of the experiment, you will be investigating the reduction of the ferricyanide anion. The purpose of this part of this experiment is to discover and develop an understanding of the properties of this reaction. This reaction can be important in the field of soil science because Iron is a comment element in soil. Iron can bind to cyanide ligands to make them less bioavailable in natural systems. The reduction of the Iron (III) complex can be represented in the following balanced half-reaction.
\[Fe(CN)_6^{-3} +e^{-}\rightarrow Fe(CN)_6^{-4} \label{1.17}\]
Four conditions will be measured in this part in order to observe the effects of concentration and counter-ion selection on the voltammogram. To begin the experiment, take note of the solutions already prepared for you. These solutions can be found in the cabinet directly under the instrument. There should be solutions of 4 mM ferricyanide in 1 M KNO3, 1 M KNO3, and 4 mM ferricyanide in 1 M Na2SO4. If these solutions are empty or missing, notify your TA.
The four conditions in this part of the experiment will use the electrodes listed in Table 1.2. The electrodes can be found in a drawer. Do not attach the electrodes until your first solution is ready.
important
For the Platinum disc electrode, note the diameter of the electrode in your lab notebook; this value will be important for your post-lab calculations.
Table 1.2: Electrodes used for Part 1 of the Experiment.
| Electrode Type | Electrode Material | Wire Color |
|---|---|---|
| Reference | Saturated Calomel Electrode (SCE) | White |
| Working | Platinum Disc | Black |
| Counter | Platinum Wire | Red |
When using these electrodes, it is important that they are clean. If the electrodes are not clean, the CV waveform produced will not be accurate. Before beginning the experiment, have your TA demonstrate the cleaning procedure for the working electrode. The cleaning procedure is as follows: Put a few drops of the cleaning solvent on a cloth. Gently rub the working electrode on the damp cloth.
The initial settings will be the same for all four conditions. These settings are listed in Table 1.3. It is important to note that the “full scale” setting can be adjusted to view your peak more clearly. An important setting not listed in Table 1.3 is the scan rate. The scan rates used will be given in the procedure sections of each condition.
| Initial Potential | +700 mV |
| Switching Potential | -200 mV |
| Final Potential | +700 mV |
| # of Segments | 2 |
| Scan Rate | 250 mV/s |
| Quiet Time | 10 seconds |
| Full Scale | 10 mA/V |
Condition 1 – 4 mM Ferricyanide in 1 M KNO3
- Fill the cell with 4 mM Ferricyanide with 1 M KNO3.
- Deoxygenate the solution for 5-10 minutes using the gas tank provided. Your TA will set the pressure gauge as needed.
- While the solution is deoxygenating, clean the electrodes, set the initial conditions in the computer software using the conditions from Table 1.2 (The first scan rate will be 250 mV/s), and prepare the dilution you will use in Condition 2 (the necessary dilution can be found in the Condition 2 section).
- Once the solution has been deoxygenated, assemble the cell, place a small stir bar into the cell, and lower the electrodes into the cell.
- Perform the scan using the procedure described in the Instrument Operation section.
- After the scan (the scan will only take a few seconds), lift the electrodes out of the solution and stir the solution for 10 seconds or until the bubbles are removed. After stirring let the solution rest for 1 minute. DO NOT stir the solution while electrodes are lowered or the scans are running, the stir bar can break the electrodes.
- Use the procedure in the Data Acquisition section to secure your data files.
- Repeat steps 5-7 at scan rates of 160, 100, 50, and 20 mV/s. Make sure to stir the solution between each scan.
- After completing your scans, ask the TA if your results look reasonable. If they do not, the electrodes may need to be cleaned using the cleaning procedure.
- Dispose of your solution in the specified waste container.
Condition 2 – 2 mM Ferricyanide in 1 M KNO3
- Dilute the 4 mM Ferricyanide with 1 M KNO3 to 2 mM Ferricyanide with 1 M KNO3. Perform the necessary dilution calculation in your laboratory notebook. This dilution needs to be done with the 1 M KNO3 solution, otherwise, the KNO3 concentration would be altered.
- Deoxygenate the solution for 5-10 minutes using the gas tank provided. Deoxygenation can begin while the scans for Condition 1 are being performed.
- Repeat steps 4-10 for this in the Condition 1 procedure for this condition running a scan with rates of 250, 160, 100, 50, and 20 mV/s.
Condition 3 – 1 M KNO3
- Fill the cell with the 1 M KNO3 solution.
- Deoxygenate the solution for 5-10 minutes using the gas tank provided. Deoxygenation can begin while the scans for Condition 2 are being performed.
- Assemble the cell with the 1 M KNO3 solution, place a small stir bar into the cell, and lower the electrodes into the cell.
- Perform the scan (scan rate = 10 mV/s) using the procedure described in the Instrument Operation section. Begin deoxygenating the solution in Condition 3.
- Do not stir the solution, no other scans will be performed for Condition 3.
- Use the procedure in the Data Acquisition section to secure your data files.
- Look at your results and try to conclude if your scan looks reasonable. If you are having a difficult time determining this, ask your TA for help.
Condition 4 – 4 mM Ferricyanide in 1 M Na2SO4
- Fill the cell with the 4 mM Ferricyanide in 1 M Na2SO4 solution.
- Deoxygenate the solution for 5-10 minutes using the gas tank provided. Deoxygenation can begin as soon as the deoxygenation of Condition 3 is complete.
- Repeat steps 4-10 in condition 1’s procedure for scan rates of 250, 100, and 50 mV/s.
- After completing the last scan, disassemble the cell, rinse the electrodes with DI water and immerse them in a cell filled with DI water. Dry the electrodes and put them away into their respective cases.
When you are finished, rinse all three electrodes with deionized water and immerse them in the cell filled with deionized water.
Part 2: The Oxidation of Acetaminophen
Tylenol is an over-the-counter pain relief medication that is commonly used in the United States. The reason that Tylenol work is because it contains the active ingredient Acetaminophen (AAPH). When someone takes Acetaminophen, also known as Paracetamol, the molecule undergoes a complete oxidation process in an aqueous solution. This oxidation reaction is the beginning of a reaction series that is demonstrated below. This is a pH-dependent process. The extent to which reaction \(\ref{1.20}\) occurs is dependent on the rate of reaction \(\ref{1.19}\).
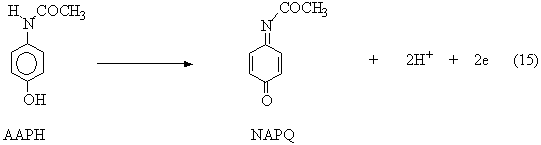 \[. \label{1.18}\]
\[. \label{1.18}\]
The product can be protonated, and then undergo the elimination of acetamide to form benzoquinone (BQ):
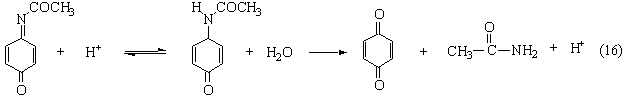 \[. \label{1.19}\]
\[. \label{1.19}\]
The rate of the first reaction is obviously pH-dependent. Finally, benzoquinone may be reduced to hydroquinone:
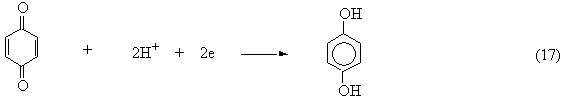 \[. \label{1.20}\]
\[. \label{1.20}\]
In this portion of the experiment, the goal is to demonstrate this mechanism and determine the best pH conditions for this reaction. After you have determined the best conditions, you will determine the concentration of AAPH in a Tylenol tablet. The electrodes used in this part are listed in Table 1.4 and the initial settings are listed in Table 1.5.
| Electrode Type | Electrode Material | Wire Color |
|---|---|---|
| Reference | Silver/Silver Chloride (Ag/AgCl) | White |
| Working | Glassy Carbon | Black |
| Counter | Platinum Wire | Red |
| Initial Potential | 0 mV |
| Switching Potential | +1000 mV |
| Final Potential | -200 mV |
| # of Segments | 2 |
| Quiet Time | 10 seconds |
| Full Scale | 10 mA/V |
Begin this part by preparing the solutions for conditions 1, 2, and 3 of pH 6, 2.2, and 1.8 respectively. Start by checking to see that there is an AAPH solution made for you. It may be in the refrigerator or in the same cabinet as the ferricyanide solution and counter-ion solutions. Once you have located the AAPH solution and three 100 mL volumetric flasks, begin by adding 5 mL of the AAPH solution to each of the volumetric flasks. The final concentration of AAPH should 3.5 mM. Fill one flask with pH 6 buffer to the line. Fill the next solution using pH 2.2 buffer and fill the last flask using 1.8 M Sulfuric Acid.
Condition 1 – pH 6
- Deoxygenate the pH 6 buffer solution for 5-10 minutes. Your TA will reset the pressure gauge if this part is performed on the second day.
- While the solution is deoxygenating, clean the electrodes with the help of your TA and set the initial conditions in the computer software using the conditions from Table 1.5 (The first scan rate will be 250 mV/s).
- Assemble the cell with the electrodes from Table 1.4 and the necessary solution. Place a small stir bar into the cell and lower the electrodes into the cell.
- Perform the scan (scan rate = 250 mV/s) using the procedure described in the Instrument Operation section. Begin deoxygenating the solution in Condition 3.
- After the scan (the scan will only take a few seconds), lift the electrodes out of the solution and stir the solution for 10 seconds or until the bubbles are removed. After stirring let the solution rest for 1 minute. DO NOT stir the solution while electrodes are lowered or the scans are running, the stir bar can break the electrodes.
- Use the procedure in the Data Acquisition section to secure your data files.
- Repeat steps 4-6 at scan rates of 100 and 40 mV/s. Make sure to stir the solution between each scan.
- Look at your results and try to conclude if your scans look reasonable. If you are having a difficult time determining this, ask your TA for help. The electrode may need to be cleaned again.
- Dispose of your solution in the specified waste container.
Condition 2 – pH 2.2
- Deoxygenate the pH 2.2 buffer solution for 5-10 minutes. This process can begin as soon as the deoxygenation of Condition 1 is complete.
- Repeat steps 3-9 in the Condition 1 section of Part 2.
Condition 3 – 1.8 M Sulfuric acid
- Deoxygenate the 1.8 M Sulfuric Acid solution for 5-10 minutes. This process can begin as soon as the deoxygenation of Condition 2 is complete.
- Repeat steps 3-9 in the Condition 1 section of Part 2.
- Do not rinse all of the electrodes and put them away, they will be used in the determination of the AAPH in the Tylenol tablet.
- As a group, determine the most appropriate condition pH condition for this reaction and the best scan rate. The best scan rate can be determined by seeing which scan gives you the clearest data. The best pH condition can be determined by picking the condition that gave the sharpest peaks.
Condition 4 – Tylenol Tablet in with the best conditions
- Weigh the Tylenol Tablet.
- Crush the tablet with a mortar and pestle into a fine powder. Transfer the fine powder into a 250 mL volumetric flask.
- Fill the volumetric flask with the appropriate pH condition that you selected.
- Deoxygenate the solution for 5-10 minutes.
- Run the scan at your chosen scan rate using the initial settings listed in Table 1.5.
- Perform the procedure in the Data Acquisition section.
- Compare the peak height in this trial to the peak height of the scan performed with the same pH condition and scan rate. Use this information to determine the approximate concentration. Use this information to develop a concentration range that can be used to develop a calibration curve.
- Dispose of your solution in the specified waste container.
Condition 5 – Calibration Curve (Solution #1)
- Make the dilution using the standard AAPH solution used in conditions 1,2, and 3 in a 100 mL volumetric flask. Fill the flask to the line with your chosen pH condition.
- Deoxygenate the solution for 5-10 minutes.
- Run the scan at the chosen scan rate using the initial settings listed in Table 1.5.
- Perform the procedure in the Data Acquisition section.
Condition 6 – Calibration Curve (Solution #2)
- Repeat the procedure from Condition 5 for the second concentration used for the calibration curve.
- After completing the last scan, disassemble the cell, rinse the electrodes with DI water and immerse them in a cell filled with DI water. Dry the electrodes and put them away into their respective cases.
Post-Lab Questions
Part 1: Reduction of the Ferricyanide Anion
**INCLUDE ALL CV WAVEFORMS**
Condition 1 – 4 mM Ferricyanide in 1 M KNO3
- Tabulate the measured values of ipc, ipa, Epc, Epa, and Epc/2 for each scan rate.
- Determine the Diffusion Coefficient (DA) for each scan rate using Equation \(\ref{1.7}\).
- Find the average, standard deviation, and relative deviation of DA for the Condition 1 scans. Discuss potential errors that could account for this deviation.
- Calculate E1/2 for each scan rate using Equation \(\ref{1.8}\).
- Test the reversibility of each scan rate using Equations \(\ref{1.11}\) and \(\ref{1.12}\). Is this reaction reversible?
Condition 2 – 2 mM Ferricyanide in 1 M KNO3
- Tabulate the measured values of ipc, ipa, Epc, Epa, and Epc/2 for each scan rate.
- Plot ipc and ipa versus v1/2 for each scan rate. Make one plot ipc versus v1/2 that includes a series from the 2 mM data and a series from the 4 mM data. Make a second plot of ipa versus v1/2 that includes a series from the 2 mM data and a series from the 4 mM data.
- Determine the Diffusion Coefficient (DA) for each scan rate using Equation \(\ref{1.7}\).
- Find the average, standard deviation, and relative deviation of DA for the Condition 2 scans. Discuss protentional errors that could account for this deviation.
- Calculate E1/2 for each scan rate using Equation \(\ref{1.8}\).
- Test the reversibility of each scan rate using Equations \(\ref{1.11}\) and \(\ref{1.12}\). Is this reaction reversible?
Condition 3 – 1 M KNO3
- Is there any current flowing through this cell? Explain.
Condition 4 – 1 M Na2SO4
- Compare your voltammograms to the voltammograms that you acquired in Condition 1. Discuss the differences.
- Does the charge of the supporting electrolyte affect the reaction?
Part 2: The Oxidation of Acetaminophen
**INCLUDE ALL CV WAVEFORMS**
- Plot ip versus v1/2. On the plot, have three data series. The series one should be the data from Condition 1, the second series is the data from Condition 2, and the third series is the data from Condition 3.
- Which system is the appropriate buffer system? Explain.
Determining the Unknown AAPH in Tylenol
- Report the mass of the Tylenol Tablet.
- Develop a calibration curve using your solutions from Condition 5 and 6. Use a third data point that uses the same pH condition and scan rate. This data point will come from the data gathered in either Condition 1, 2, or 3.
- Use the calibration curve to determine the concentration of AAPH and report this value.
- Calculate and report the mass of AAPH in the tablet.
- Calculate and report the mass percent. Discuss any error in scans or in the determination for this value.
Outside Links
- http://www.chem.uoa.gr/applets/Apple...l_Diffus2.html
- http://pubs.acs.org/doi/abs/10.1021/j100661a017
References
- Heineman, W. R.; Kissinger, P. T. Large-Amplitude Controled-Potential Techniques. In Laboratory Techniques in Electroanalytical Chemistry; Kissinger, P. T., Heineman, W. R., Eds.; Monographs in Electroanalytical Chemistry and Electrochemistry; Marcel Dekker: New York, NY, 1984; pp 51-127.
- Bard, A. J.; Faulkner, L. R. Electrochemical Methods; Wiley: New York, NY, 1980; Chapter 6.
- Compton, R. G.; Banks, C. E. Understanding Voltammetry; World Scientific: Hackensack, NJ, 2007.
- Skoog, D., Holler, F. J., & Crouch, S. R. (2017). Principles of Instrumental Analysis (Seventh ed.). Boston, MA: Cengage Learning.
-
Elgrishi, N.; Rountree, K.J.; Brian D. McCarthy, B.D.; et al. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem, Ed. 2018 95 (2), 197-206.