
12.6: Other Forms of Chromatography
- Page ID
- 220768

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)At the beginning of Section 12.5, we noted that there are several different types of solute/stationary phase interactions in liquid chromatography, but limited our discussion to liquid–liquid chromatography. In this section we turn our attention to liquid chromatography techniques in which partitioning occurs by liquid–solid adsorption, ion-exchange, and size exclusion.
Liquid-Solid Chromatography
In liquid–solid adsorption chromatography (LSC) the column packing also serves as the stationary phase. In Tswett’s original work the stationary phase was finely divided CaCO3, but modern columns employ porous 3–10 μm particles of silica or alumina. Because the stationary phase is polar, the mobile phase usually is a nonpolar or a moderately polar solvent. Typical mobile phases include hexane, isooctane, and methylene chloride. The usual order of elution—from shorter to longer retention times—is
olefins < aromatic hydrocarbons < ethers < esters, aldehydes, ketones < alcohols, amines < amide < carboxylic acids
Nonpolar stationary phases, such as charcoal-based absorbents, also are used. For most samples, liquid–solid chromatography does not offer any special advantages over liquid–liquid chromatography. One exception is the analysis of isomers, where LSC excels.
Ion-Exchange Chromatography
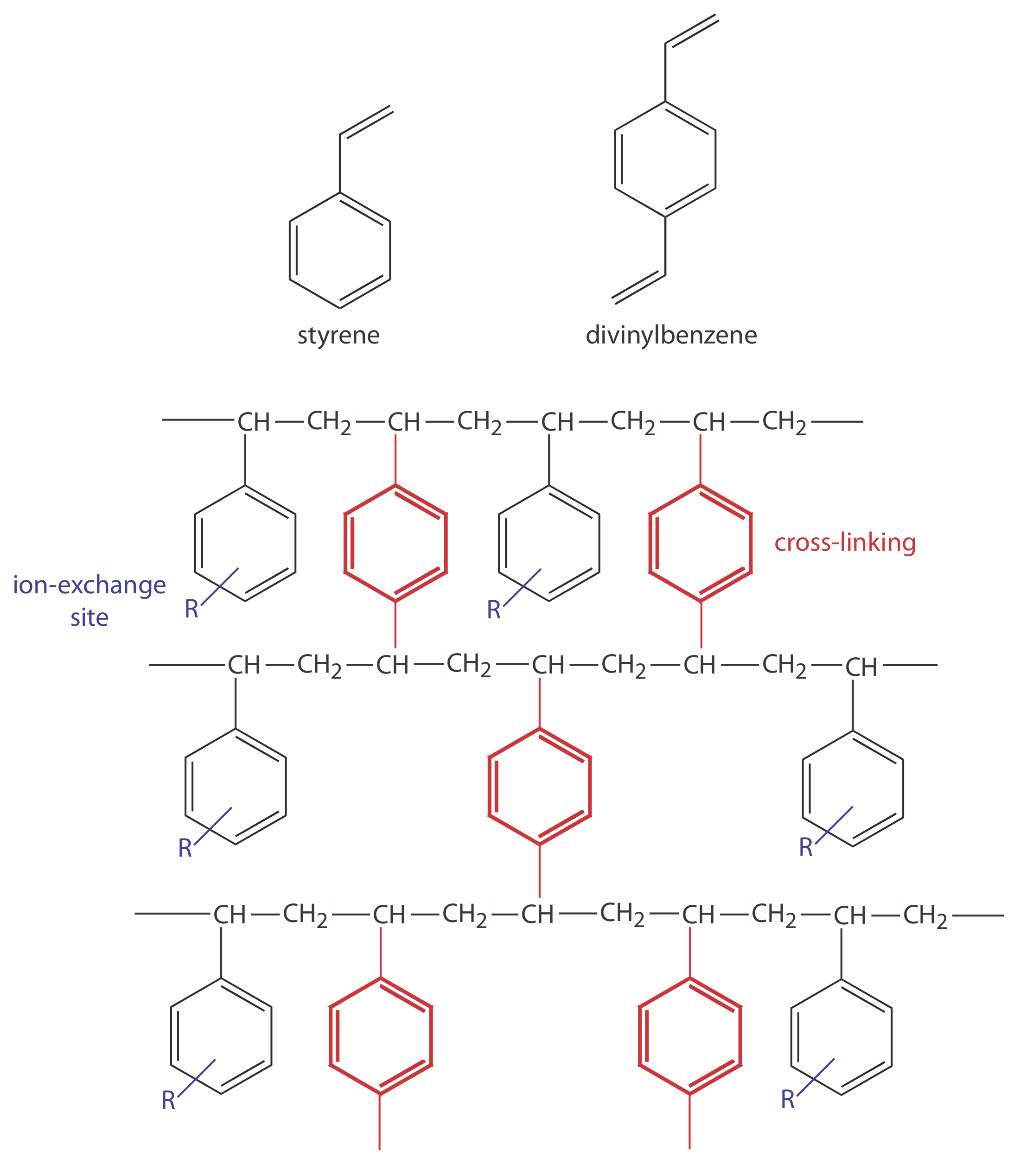
In ion-exchange chromatography (IEC) the stationary phase is a cross-linked polymer resin, usually divinylbenzene cross-linked polystyrene, with covalently attached ionic functional groups (see Figure \(\PageIndex{1}\) and Table \(\PageIndex{1}\)). The counterions to these fixed charges are mobile and are displaced by ions that compete more favorably for the exchange sites. Ion-exchange resins are divided into four categories: strong acid cation exchangers; weak acid cation exchangers; strong base anion exchangers; and weak base anion exchangers.
 |
 |
Figure \(\PageIndex{1}\). Structures of styrene, divinylbenzene, and a styrene–divinylbenzene co-polymer modified for use as an ion-exchange resin are shown on the left. The ion-exchange sites, indicated by R and shown in blue, are mostly in the para position and are not necessarily bound to all styrene units. The cross-linking is shown in red. The photo on the right shows an example of the polymer beads. These beads are approximately 0.30–0.85 mm in diameter. Resins for use in ion-exchange chromatography typically are 5–11 μm in diameter.
Strong acid cation exchangers include a sulfonic acid functional group that retains it anionic form—and thus its capacity for ion-exchange—in strongly acidic solutions. The functional groups for a weak acid cation exchanger, on the other hand, are fully protonated at pH levels less then 4 and lose their exchange capacity. The strong base anion exchangers include a quaternary amine, which retains a positive charge even in strongly basic solutions. Weak base anion exchangers remain protonated only at pH levels that are moderately basic. Under more basic conditions a weak base anion exchanger loses a proton and its exchange capacity.
The ion-exchange reaction of a monovalent cation, M+, exchange site is
\[-\mathrm{SO}_{3}^{-} \mathrm{H}^{+}(s)+\mathrm{M}^{+}(a q)\rightleftharpoons-\mathrm{SO}_{3}^{-} \mathrm{M}^{+}(s)+\mathrm{H}^{+}(a q) \nonumber\]
The equilibrium constant for this ion-exchange reaction, which we call the selectivity coefficient, K, is
\[K=\frac{\left\{-\mathrm{SO}_{3}^{-} \mathrm{M}^{+}\right\}\left[\mathrm{H}^{+}\right]}{\left\{-\mathrm{SO}_{3}^{-} \mathrm{H}^{+}\right\}\left[\mathrm{M}^{+}\right]} \label{12.1}\]
where we use curly brackets, { }, to indicate a surface concentration instead of a solution concentration.
We don’t usually think about a solid’s concentration. There is a good reason for this. In most cases, a solid’s concentration is a constant. If you break a piece of chalk into two parts, for example, the mass and the volume of each piece retains the same proportional relationship as in the original piece of chalk. When we consider an ion binding to a reactive site on the solid’s surface, however, the fraction of sites that are bound, and thus the concentration of bound sites, can take on any value between 0 and some maximum value that is proportional to the density of reactive sites.
Rearranging equation \ref{12.1} shows us that the distribution ratio, D, for the exchange reaction
\[D=\frac{\text { amount of } \mathrm{M}^{+} \text { in the stationary phase }}{\text { amount of } \mathrm{M}^{+} \text { in the mobile phase }} \nonumber\]
\[D=\frac{\left\{-\mathrm{SO}_{3}^{-} \mathrm{M}^{+}\right\}}{\left[\mathrm{M}^{+}\right]}=K \times \frac{\left\{-\mathrm{SO}_{3}^{-} \mathrm{H}^{+}\right\}}{\left[\mathrm{H}^{+}\right]} \label{12.2}\]
is a function of the concentration of H+ and, therefore, the pH of the mobile phase.
An ion-exchange resin’s selectivity is somewhat dependent on whether it includes strong or weak exchange sites and on the extent of cross-linking. The latter is particularly important as it controls the resin’s permeability, and, therefore, the accessibility of exchange sites. An approximate order of selectivity for a typical strong acid cation exchange resin, in order of decreasing D, is
Al3+ > Ba2+ > Pb2+ > Ca2+ > Ni2+ > Cd2+ > Cu2+ > Co2+ > Zn2+ > Mg2+ > Ag+ > K+ > \(\text{NH}_4^+\) > Na+ > H+ > Li+
Note that highly charged cations bind more strongly than cations of lower charge, and that for cations of similar charge, those with a smaller hydrated radius (see Table 6.9.1 in Chapter 6), or that are more polarizable, bind more strongly. For a strong base anion exchanger the general elution order is
\(\text{SO}_4^{2-}\) > I– > \(\text{HSO}_4^-\) > \(\text{NO}_3^-\) > Br– > \(\text{NO}_2^-\) > Cl– > \(\text{HCO}_3^-\) > CH3COO– > OH– > F–
Anions of higher charge and of smaller hydrated radius bind more strongly than anions with a lower charge and a larger hydrated radius.
The mobile phase in IEC usually is an aqueous buffer, the pH and ionic composition of which determines a solute’s retention time. Gradient elutions are possible in which the mobile phase’s ionic strength or pH is changed with time. For example, an IEC separation of cations might use a dilute solution of HCl as the mobile phase. Increasing the concentration of HCl speeds the elution rate for more strongly retained cations because the higher concentration of H+ allows it to compete more successfully for the ion-exchange sites.
From equation \ref{12.2}, a cation’s distribution ratio, D, becomes smaller when the concentration of H+ in the mobile phase increases.
An ion-exchange resin is incorporated into an HPLC column either as 5–11 μm porous polymer beads or by coating the resin on porous silica particles. Columns typically are 250 mm in length with internal diameters ranging from 2–5 mm.
Measuring the conductivity of the mobile phase as it elutes from the column serves as a universal detector for cationic and anionic analytes. Because the mobile phase contains a high concentration of ions—a mobile phase of dilute HCl, for example, contains significant concentrations of H+ and Cl–—we need a method for detecting the analytes in the presence of a significant background conductivity.
To minimize the mobile phase’s contribution to conductivity, an ion-suppressor column is placed between the analytical column and the detector. This column selectively removes mobile phase ions without removing solute ions. For example, in cation-exchange chromatography using a dilute solution of HCl as the mobile phase, the suppressor column contains a strong base anion-exchange resin. The exchange reaction
\[\mathrm{H}^{+}(a q)+\mathrm{Cl}^{-}(a q)+\mathrm{Resin}^{+} \mathrm{OH}^{-}(s)\rightleftharpoons\operatorname{Resin}^{+} \mathrm{Cl}^{-}(s)+\mathrm{H}_{2} \mathrm{O}(l ) \nonumber\]
replaces the mobile phase ions H+ and Cl– with H2O. A similar process is used in anion-exchange chromatography where the suppressor column contains a cation-exchange resin. If the mobile phase is a solution of Na2CO3, the exchange reaction
\[2 \mathrm{Na}^{+}(a q)+\mathrm{CO}_{3}^{2-}(a q)+2 \operatorname{Resin}^{-} \mathrm{H}^{+}(s)\rightleftharpoons2 \operatorname{Resin}^{-} \mathrm{Na}^{+}(s)+\mathrm{H}_{2} \mathrm{CO}_{3}(a q) \nonumber\]
replaces a strong electrolyte, Na2CO3, with a weak electrolyte, H2CO3.
Ion-suppression is necessary when the mobile phase contains a high concentration of ions. Single-column ion chromatography, in which an ion-suppressor column is not needed, is possible if the concentration of ions in the mobile phase is small. Typically the stationary phase is a resin with a low capacity for ion-exchange and the mobile phase is a very dilute solution of methane sulfonic acid for cationic analytes, or potassium benzoate or potassium hydrogen phthalate for anionic analytes. Because the background conductivity is sufficiently small, it is possible to monitor a change in conductivity as the analytes elute from the column.
A UV/Vis absorbance detector can be used if the analytes absorb ultraviolet or visible radiation. Alternatively, we can detect indirectly analytes that do not absorb in the UV/Vis if the mobile phase contains a UV/Vis absorbing species. In this case, when a solute band passes through the detector, a decrease in absorbance is measured at the detector.
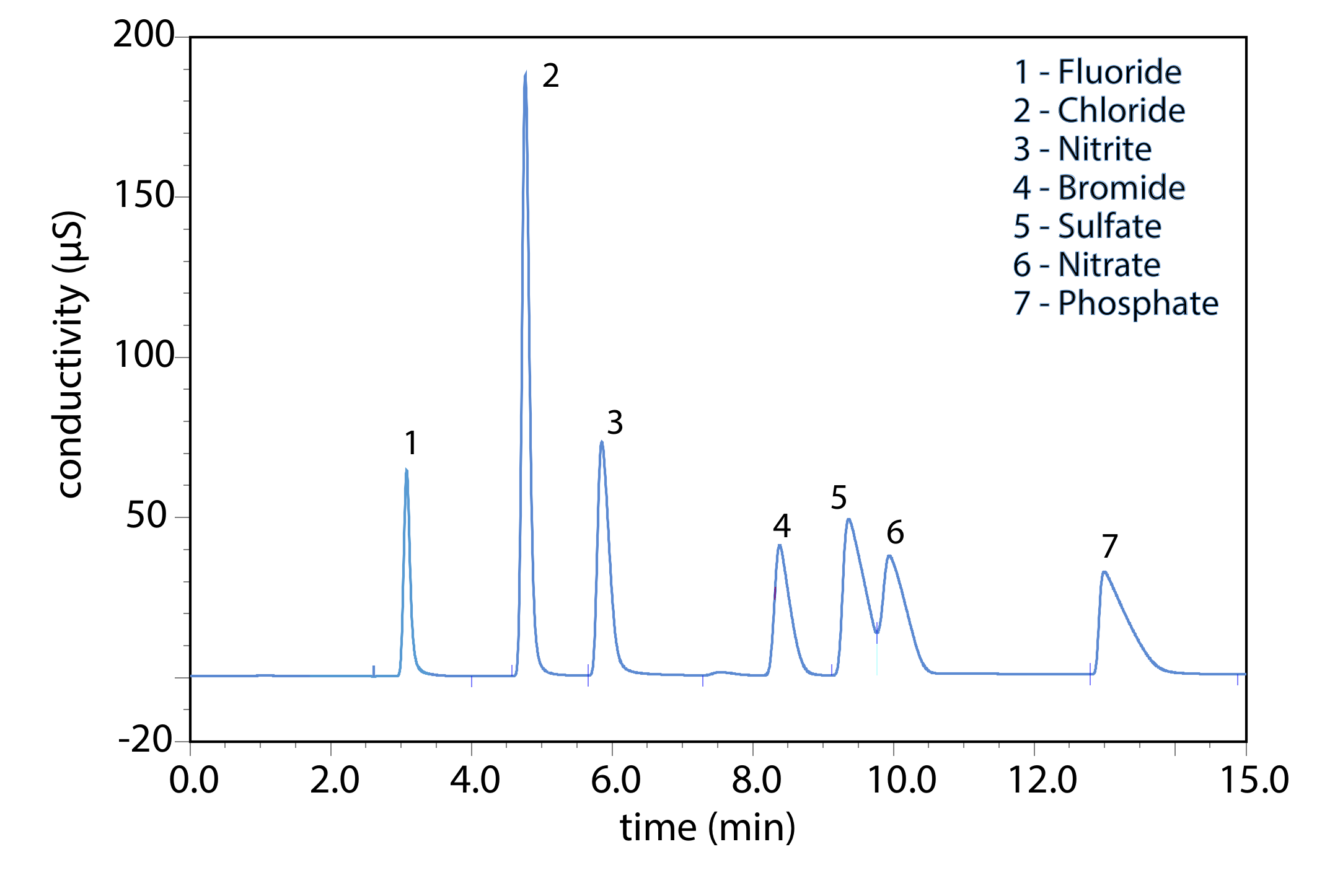
Ion-exchange chromatography is an important technique for the analysis of anions and cations in water. For example, an ion-exchange chromatographic analysis for the anions F–, Cl–, Br–, \(\text{NO}_2^-\) , \(\text{NO}_3^-\) , \(\text{PO}_4^{3-}\) , and \(\text{SO}_4^{2-}\) takes approximately 15 minutes (Figure \(\PageIndex{2}\)). A complete analysis of the same set of anions by a combination of potentiometry and spectrophotometry requires 1–2 days. Ion-exchange chromatography also is used for the analysis of proteins, amino acids, sugars, nucleotides, pharmaceuticals, consumer products, and clinical samples.
Size-Exclusion Chromatography
We have considered two classes of micron-sized stationary phases in this section: silica particles and cross-linked polymer resin beads. Both materials are porous, with pore sizes ranging from approximately 5–400 nm for silica particles, and from 5 nm to 100 μm for divinylbenzene cross-linked polystyrene resins. In size-exclusion chromatography—which also is known by the terms molecular-exclusion or gel permeation chromatography—the separation of solutes depends upon their ability to enter into the pores of the stationary phase. Smaller solutes spend proportionally more time within the pores and take longer to elute from the column.
A stationary phase’s size selectivity extends over a finite range. All solutes significantly smaller than the pores move through the column’s entire volume and elute simultaneously, with a retention volume, Vr, of
\[V_{r}=V_{i}+V_{o} \label{12.3}\]
where Vi is the volume of mobile phase occupying the stationary phase’s pore space and Vo is volume of mobile phase in the remainder of the column. The largest solute for which equation \ref{12.3} holds is the column’s inclusion limit, or permeation limit. Those solutes too large to enter the pores elute simultaneously with an retention volume of
\[V_{r} = V_{o} \label{12.4}\]
Equation \ref{12.4} defines the column’s exclusion limit.
For a solute whose size is between the inclusion limit and the exclusion limit, the amount of time it spends in the stationary phase’s pores is proportional to its size. The retention volume for these solutes is
\[V_{r}=DV_{i}+V_{o} \label{12.5}\]
where D is the solute’s distribution ratio, which ranges from 0 at the exclusion limit to 1 at the inclusion limit. Equation \ref{12.5} assumes that size-exclusion is the only interaction between the solute and the stationary phase that affects the separation. For this reason, stationary phases using silica particles are deactivated as described earlier, and polymer resins are synthesized without exchange sites.
Size-exclusion chromatography provides a rapid means for separating larger molecules, including polymers and biomolecules. A stationary phase for proteins that consists of particles with 30 nm pores has an inclusion limit of 7500 g/mol and an exclusion limit of \(1.2 \times 10^6\) g/mol. Mixtures of proteins that span a wider range of molecular weights are separated by joining together in series several columns with different inclusion and exclusion limits.
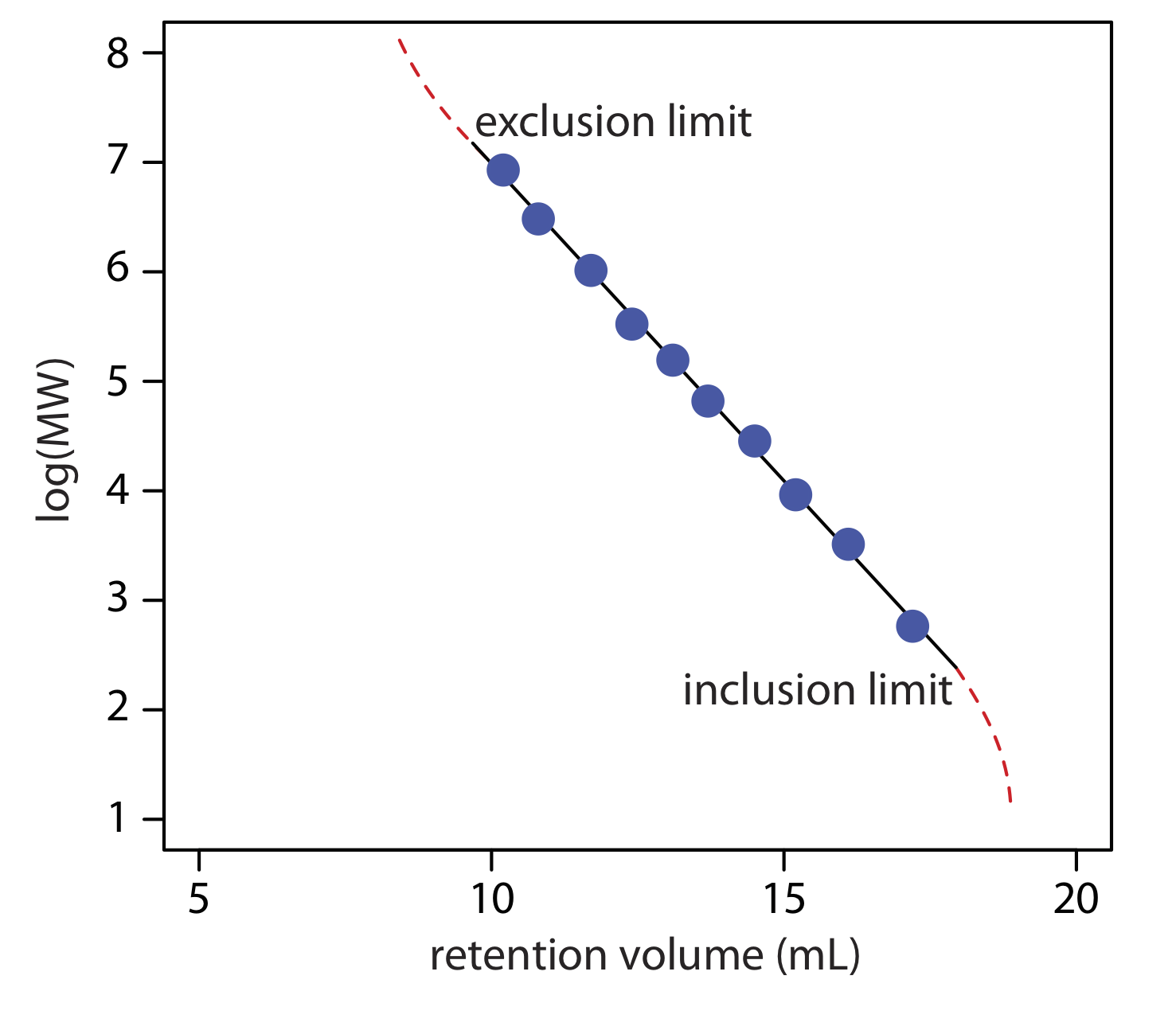
Another important application of size-exclusion chromatography is the estimation of a solute’s molecular weight (MW). Calibration curves are prepared using a series of standards of known molecular weight and measuring each standard’s retention volume. As shown in Figure \(\PageIndex{3}\), a plot of log(MW) versus Vr is roughly linear between the exclusion limit and the inclusion limit. Because a solute’s retention volume is influenced by both its size and its shape, a reasonably accurate estimation of molecular weight is possible only if the standards are chosen carefully to minimize the effect of shape.
Size-exclusion chromatography is carried out using conventional HPLC instrumentation, replacing the HPLC column with an appropriate size-exclusion column. A UV/Vis detector is the most common means for obtaining the chromatogram.
Supercritical Fluid Chromatography
Although there are many analytical applications of gas chromatography and liquid chromatography, they can not separate and analyze all types of samples. Capillary column GC separates complex mixtures with excellent resolution and short analysis times. Its application is limited, however, to volatile analytes or to analytes made volatile by a suitable derivatization reaction. Liquid chromatography separates a wider range of solutes than GC, but the most common detectors—UV, fluorescence, and electrochemical— have poorer detection limits and smaller linear ranges than GC detectors, and are not as universal in their selectivity.
For some samples, supercritical fluid chromatography (SFC) provides a useful alternative to gas chromatography and liquid chromatography. The mobile phase in supercritical fluid chromatography is a gas held at a temperature and pressure that exceeds its critical point (Figure \(\PageIndex{4}\)). Under these conditions the mobile phase is neither a gas nor a liquid. Instead, the mobile phase is a supercritical fluid.
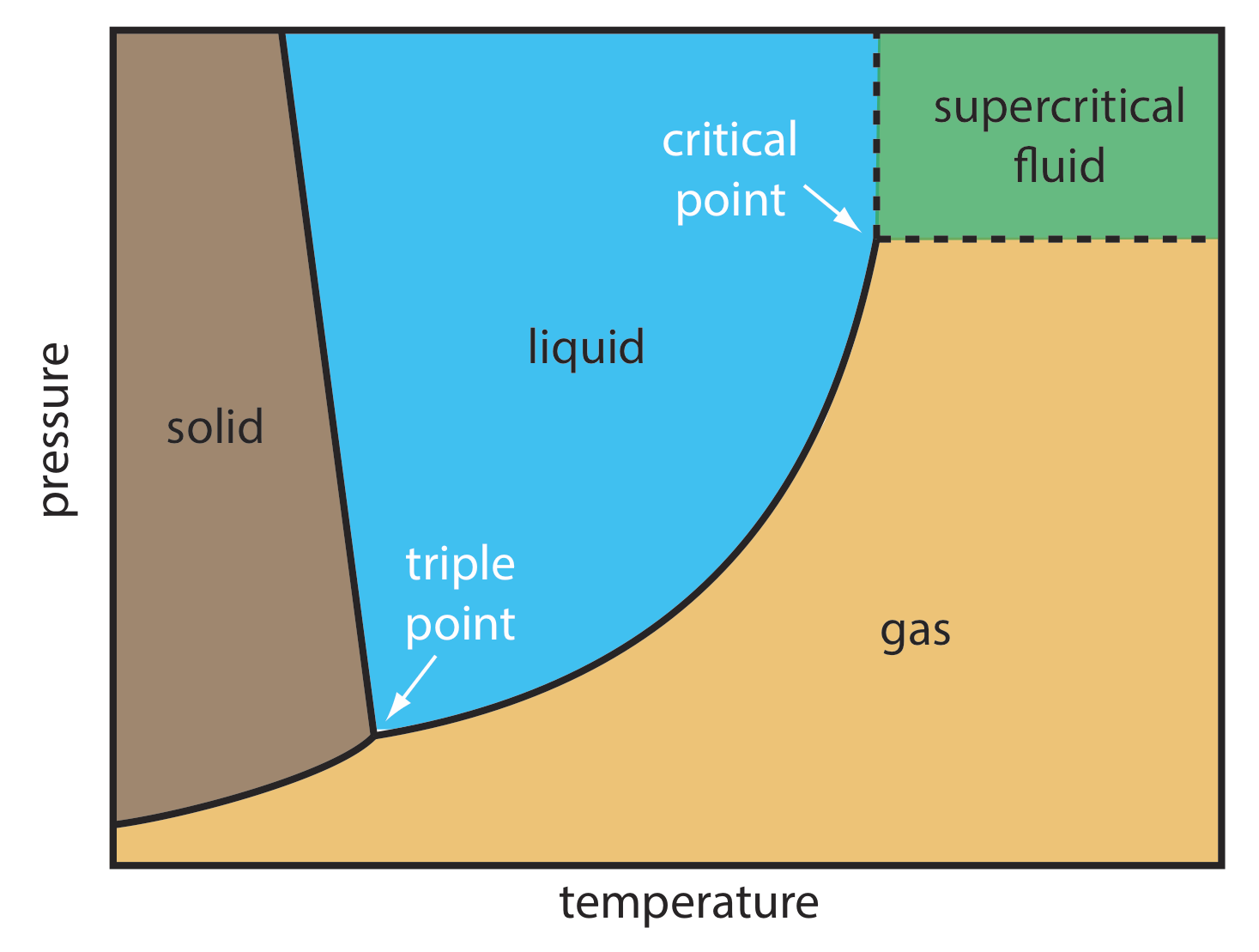
Some properties of a supercritical fluid, as shown in Table \(\PageIndex{2}\), are similar to a gas; other properties, however, are similar to a liquid. The viscosity of a supercritical fluid, for example, is similar to a gas, which means we can move a supercritical fluid through a capillary column or a packed column without the high pressures needed in HPLC. Analysis time and resolution, although not as good as in GC, usually are better than in conventional HPLC. The density of a supercritical fluid, on the other hand, is much closer to that of a liquid, which explains why supercritical fluids are good solvents. In terms of its separation power, a mobile phase in SFC behaves more like the liquid mobile phase in HPLC than the gaseous mobile phase in GC.
The most common mobile phase for supercritical fluid chromatography is CO2. Its low critical temperature of 31.1oC and its low critical pressure of 72.9 atm are relatively easy to achieve and maintain. Although supercritical CO2 is a good solvent for nonpolar organics, it is less useful for polar solutes. The addition of an organic modifier, such as methanol, improves the mobile phase’s elution strength. Other common mobile phases and their critical temperatures and pressures are listed in Table \(\PageIndex{3}\).
The instrumentation for supercritical fluid chromatography essentially is the same as that for a standard HPLC. The only important additions are a heated oven for the column and a pressure restrictor downstream from the column to maintain the critical pressure. Gradient elutions are accomplished by changing the applied pressure over time. The resulting change in the mobile phase’s density affects its solvent strength. Detection is accomplished using standard GC detectors or HPLC detectors. Supercritical fluid chromatography has many applications in the analysis of polymers, fossil fuels, waxes, drugs, and food products.