
Schmidt Reaction
- Page ID
- 65181

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The Schmidt reaction is the reaction of hydrazoic acid or an alkyl azide with a carbonyl compound, alkene, or alcohol, often in the presence of a Brønsted or Lewis acid. Although the family of Schmidt reactions includes a number of variants, they all result in the migration of a substituent from carbon to nitrogen with loss of a molecule of dinitrogen. This reaction has considerable utility for the synthesis of hindered or cyclic amides and amines.[1]
Introduction
Azides are nucleophilic at their terminal nitrogen atoms, and may add to suitably activated electrophiles in the presence of a Brønsted or Lewis acid. Upon addition, the newly bound nitrogen atom becomes electron-deficient and is subject to 1,2-migration of a carbon or hydrogen substituent with loss of a molecule of dinitrogen. Historically, carbonyl compounds were the first electrophiles successfully employed in this context.[2] Since the initial discovery of the Schmidt reaction, many variants employing alternative electrophiles and hydrazoic acid have been developed (Eq. 1). Related reactions of alkyl azides may yield substituted amides, lactams, or amines (after reduction of iminium ions). However, the scope of alkyl azides in the Schmidt reaction is limited compared to hydrazoic acid.[3]
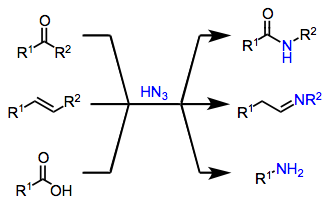 (1)
(1)
Generally, intramolecular Schmidt reactions are more useful than their intermolecular counterparts, which are limited by poor site selectivity and sensitivity to steric hindrance. The Schmidt reaction of carboxylic acids, which produces amines, is in direct competition with the milder Curtius rearrangement,[4] and is rarely used in practice. Nonetheless, the Schmidt reaction has been applied extensively for the synthesis of medium-sized lactams and hindered amides. For these applications, the Schmidt reaction exhibits advantageous site selectivity and atom economy.
Mechanism and Stereochemistry
Reactions of Ketones
The mechanisms of reactions of ketones with hydrazoic acid begin with addition of HN3 to the carbonyl group to form adizohydrin intermediate I. At this stage, one can envision two mechanistic pathways that lead to amides. In a process analogous to the mechanism of the Baeyer-Villiger reaction (Eq. 2, blue pathway), the azidohydrin may rearrange to afford amides directly after loss of a proton.[5] A second possibility (Eq. 2, green pathway) is analogous to the mechanism of the Beckmann rearrangement. Elimination of water from the azidohydrin affords isomeric diazoiminium ions II and III, which undergo synchronous, antiperiplanar 1,2-rearrangement to yield nitrilium ions IVand V. Subsequent addition of water and tautomerization lead to the amide products.[6] The observation of tetrazole side products at high concentrations of hydrazoic acid[7] and similar product ratios in conceptually related Beckmann and Schmidt reactions[8] provide evidence of the Beckmann-type mechanism in reactions of hydrazoic acid. However, it should be noted that alkyl azides must react via the Baeyer-Villiger mechanism to avoid the formation of adjacent positive charges.
(2)
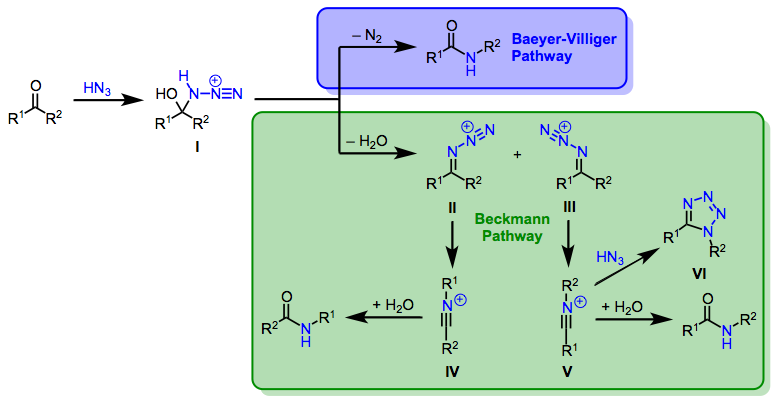
The product of the migration step is determined by the configuration of the diazoiminium ion, and it has thus been proposed that the observed amide product ratio is directly related to the ratio of diazoiminium ions. In support of this idea, calculations have shown that the barrier to direct conversion between II and III is quite high.[9] However, II and III may interconvert rapidly via the addition of water followed by elimination, and a Curtin-Hammett situation cannot be ruled out. Under Curtin-Hammett conditions, the amide product ratio is not determined by the ratio of diazoiminium ions, but by their relative rates of conversion to nitrilium ions. In any case, preferential migration of the larger R group is overwhelmingly observed, suggesting the predominant intermediacy of the less hindered diazoiminium ion.[8] Stereogenic centers migrate with retention of configuration.[10]
Reactions of Carboxylic Acids
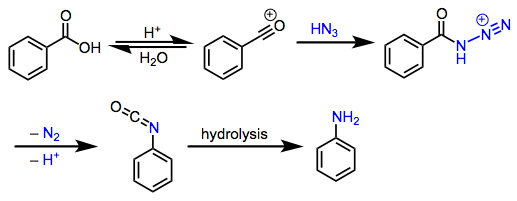
The mechanism of the reaction of carboxylic acids with hydrazoic acid is well understood. Initial formation of an acylium ion is followed by the addition of hydrazoic acid to form an acyl azide intermediate. The only possible migration event produces an isocyanate, which undergoes hydrolysis to yield an amine and carbon dioxide (Eq. 3).[1]
(3)
Reactions of Other Electrophiles
Electrophiles capable of forming carbocationic intermediates may also participate in the Schmidt reaction. After nucleophilic addition of hydrazoic acid to the carbocation, alkyl or aryl group migration affords an imine (Eq. 2). Subsequent hydrolysis and tautomerization of the imine are both possible.[11]
(4)
The relative electron density of the R groups is likely the primary determinant of site selectivity in these Baeyer-Villiger-type reactions. More electron-rich, nucleophilic R groups migrate to a greater extent.[12]
Enantioselective Variants
The Schmidt reaction has been applied for the desymmetrization of symmetric ketones containing enantiotopic α-carbon atoms. Employing a chiral hydroxyalkyl azide resulted in highly diastereoselective migration, and subsequent removal of the nitrogen substituent produced the lactam as a single enantiomer.[13]
(5)
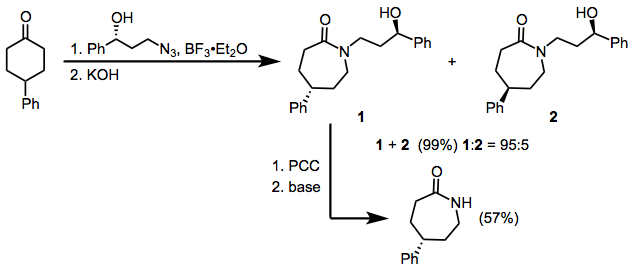
Scope and Limitations
Reactions of Hydrazoic Acid
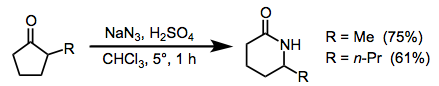
The Schmidt reaction is most commonly used to convert differentially substituted ketones to amides or lactams. The most common problems with this reaction are site selectivity and tetrazole formation, although the latter can be controlled by changing reaction conditions. Steric hindrance to nucleophilic addition lowers yields somewhat, but site selectivity is improved when the substituents of the carbonyl are of different sizes. The less substituted carbon rarely migrates (Eq. 6).[14]
(6)
Schmidt reactions of aldehydes are often problematic because of amide and nitrile formation (C-migration and H-migration, respectively).[15]
Aromatic groups often migrate in preference to alkyl groups in reactions of ketones, particularly if the ring is electron rich (Eq. 7).[16] The behavior of heteroaromatic ketones is more difficult to predict.[17]
(7)
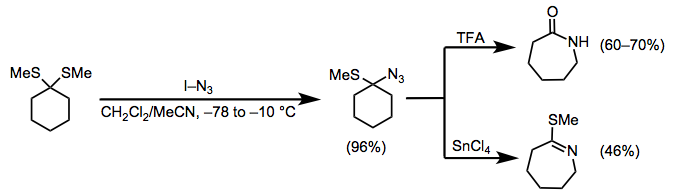
A variety of alternative electrophiles react with hydrazoic acid to afford products of Schmidt-type reactions. For instance, dithioketals activated by iodoazide form α-azido sulfides, which may be converted to amides via acidic hydrolysis or to thioimino ethers with stannic tetrachloride (Eq. 8).[18]
(8)
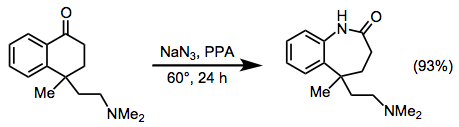
Reactions of Alkyl Azides
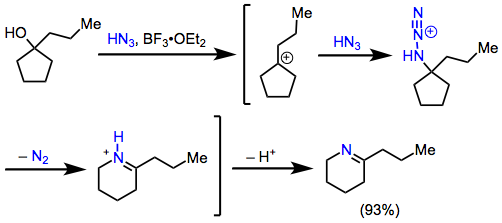
The scope of reactions of alkyl azides is generally more limited than the scope of reactions of hydrazoic acid. Intermolecular Schmidt reactions of alkyl azides are plagued by poor site selectivity and strong sensitivity to steric effects. However, intramolecular reactions of alkyl azides are among the most synthetically useful applications of the Schmidt reaction. The range of ketones that engage in the intramolecular Schmidt reactions is exceptionally broad. For these reactions, the azide and carbonyl carbon must be separated by four or five atoms to facilitate cyclization, with four preferred (Eq. 9). Substrates containing five-carbon tethers react only in the presence of relatively strong Lewis acids.[19]
(9)
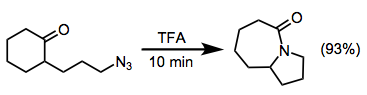
One useful intermolecular method involves the use of hydroxyalkyl azides, whose pendant hydroxy groups may attack the intermediate oxocarbenium ion present after migration to form iminium ethers. Subsequent basic hydrolysis leads to the hydroxyalkyl-substituted amide, and nucleophiles other than hydroxide have been employed with some success.[20]
(10)
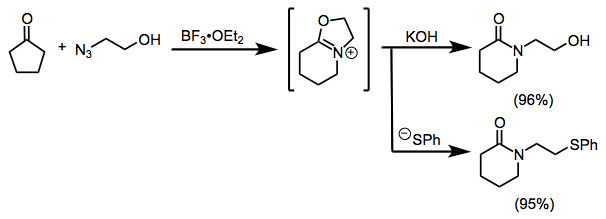
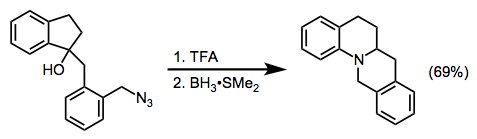
Many Schmidt reactions of cationic electrophiles are subject to carbocationic rearrangements. However, a judicious choice of reaction conditions (and utilizing an intramolecularly bound azide) can steer the reaction toward a single product (Eq. 11).[21]. Intermolecular reactions of benzylic and tertiary cations with alkyl azides are also known.[22]
(11)
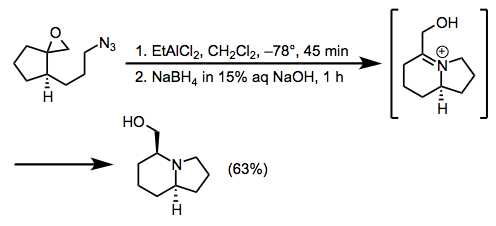
Epoxides are a useful class of cation equivalents that react rapidly with alkyl azides in the presence of a Lewis acid to afford hydroxymethyl iminium ions. Subsequent reduction with sodium borohydride yields the corresponding amine (Eq. 12).[23]
(12)
Synthetic Applications
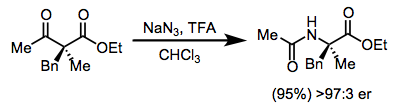
The Schmidt reaction has been applied to the synthesis of disubstituted amino acids. After asymmetric alkylation of a β-ketoester, treatment with trifluoroacetic acid (TFA) and sodium azide yields the N-acetyl amino ester, which may be converted to the corresponding amino acid through functional group manipulations (Eq. 13).[24]
(13)
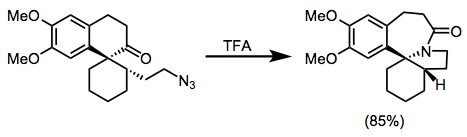
The Schmidt reaction has also been applied extensively to access crowded or otherwise difficult amines and amides in alkaloid natural products. Efforts toward the homoerythrina spirocyclic ring system contain a nice application of the intramolecular Schmidt reaction of alkyl azides. The use of Beckmann conditions and hydrazoic acid both failed for the sterically hindered ketone in Eq. 14; however, an appropriately positioned alkyl azide was smoothly converted to the lactam in the presence of TFA.[25]
(14)
Comparison to Other Methods
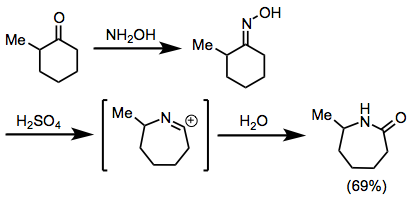
The Beckmann rearrangement is conceptually very similar to the Schmidt reaction, but involves the discrete formation of an oxime (analogous to the posited diazoiminium intermediate of the Schmidt reaction) prior to the rearrangement step. Like the Schmidt reaction, the site selectivity of the Beckmann rearrangement is generally controlled by stereoelectronic factors and the relative prevalence or reactivity of geometric isomers. The site selectivity of related Schmidt and Beckmann reactions are generally similar (migration of the larger group is favored, for instance).[26]
(15)
The sense of site selectivity of the photochemical oxaziridine rearrangement is opposite that of the Beckmann and Schmidt reactions. In fact, this reaction is one of a select few that result in the selective insertion of nitrogen into the less hindered of two carbon-carbon bonds (Eq. 16).[27]
(16)
Although Schmidt reactions of carboxylic acids are certainly possible and have been studied in detail, the Curtius rearrangement of carboxylic acids to isocyanates is a much more mild and practical method for the synthesis of amines from carboxylic acids (Eq. 17).[28] In addition, the initial isocyanate products can easily be converted to urethanes and ureas through treatment with alcohols or amines, an impossible transformation under the conditions of the Schmidt reaction.
(17)
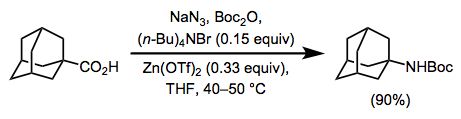
Experimental Conditions and Procedure
Typical Conditions
Hydrazoic acid is typically prepared in situ for immediate use via treatment of sodium azide with hydrochloric or sulfuric acid. In some cases an additional Lewis acid is added. Hydrazoic acid is both explosive and toxic, and should be handled with extreme care in a well-ventilated fume hood with a safety shield. Alkyl azides are in many cases explosive and toxic as well (particularly alkyl azides of low molecular weight). While intermolecular reactions typically employ titanium tetrachloride as a Lewis acid, either Lewis or Brønsted acids (most commonly, trifluoroacetic acid) may be used for intramolecular reactions.
An important caution concerns the use of sodium azide in methylene chloride solutions for the synthesis of alkyl azides. The formation of extremely explosive diazidomethane under these conditions has been observed.[29]Alternative methods for synthesizing alkyl azides from alcohols using Mitsunobu conditions[30] and from alkyl halides using azidotrimethylsilane[31] are available.
Example Procedure
(18)
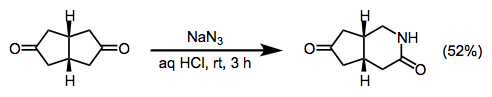
Sodium azide (0.60 g, 9.1 mmol) was added portionwise over 20 min to a solution of cis-bicyclo[3.3.0]octane-3,7-dione (1.0 g, 7.2 mmol) in 36% aqueous HCl (20 mL), while keeping the temperature below 35 °C. The mixture was stirred for 3 h at rt and then brought to pH 10 with 20% aqueous NaOH at 0 °C. Precipitated NaCl was filtered off and the aqueous layer was extracted continuously with CHCl3 for 48 h. Drying the organic phase over MgSO4, followed by removal of solvent, furnished a residue that was separated on a silica gel column (30 g). Elution with CH2Cl2/acetone (85:15) gave unreacted diketone (240 mg) and the desired product (576 mg, 52%): mp 120–122 °C; 1H NMR (CDCl3) δ 2.10–2.30 (m, 3H), 2.40–2.90 (m, 5H), 3.21 (ddd, J = 3.0, 6.8, 13.0 Hz, 1H), 3.52 (ddd, J = 3.5, 5.8, 13.0 Hz, 1H), 6.3 (br s, 1H); 13C NMR (D2O) δ 30.7 (d), 31.3 (d), 31.9 (t), 40.9 (t), 41.9 (t), 43.6 (t), 175.4 (s), 223.7 (s); IR (neat) 3246, 2935, 1735, 1667, 1638 cm–1; MS m/z: M+ 153 (100), 125 (10), 112 (33), 96 (35), 82 (53), 68 (33), 54, (82), 41 (78); Anal. Calcd for C8H11NO2: C, 62.73; H, 7.24; N, 9.14. Found: C, 62.76; H, 7.21; N, 9.11.
References
- ↑ a b Wrobleski, A.; Coombs, T. C.; Huh, C. W.; Li, S.-W.; Aubé, J. Org. React. 2011, 78, 1. doi: (10.1002/0471264180.or078.01)
- ↑ Wolff, H. Org. React. 1946, 3, 307. doi: (10.1002/0471264180.or003.08)
- ↑ Aubé, J.; Milligan, G. L.; Mossman, C. J. J. Org. Chem. 1992, 57, 1635.
- ↑ Braese, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188.
- ↑ Krow, G. R. Org. React. 1993, 43, 251. doi: (10.1002/0471264180.or043.03)
- ↑ Smith, P. A. S. J. Am. Chem. Soc. 1948, 70, 320.
- ↑ Amyes, T. L.; Richard, J. P. J. Am. Chem. Soc. 1991, 113, 1867.
- ↑ a b Smith, P. A. S.; Ashby, B. J. Am. Chem. Soc. 1950, 72, 2503.
- ↑ Bach, R. D.; Wolber, G. J. J. Org. Chem. 1982, 47, 239.
- ↑ Siddiqui, A. H.; Afzal, M. N.; Baig, M. H.; Lingam, G. V.; Rao, N. S. Indian J. Chem. 1984, 23B, 1108.
- ↑ Astier, A.; Plat, M. M. Tetrahedron Lett. 1978, 19, 2051.
- ↑ Nielsen, D. R.; McEwen, W. E. J. Am. Chem. Soc. 1954, 76, 4042.
- ↑ Gracias, V.; Milligan, G. L.; Aubé, J. J. Am. Chem. Soc. 1995, 117, 8047.
- ↑ Krow, G. R. Tetrahedron 1981, 37, 1283.
- ↑ Boyer, J. H.; Hamer, J. J. Am. Chem. Soc. 1955, 77, 951.
- ↑ Carr, D.; Iddon, B.; Suschitzky, H.; Parfitt, R. T. J. Chem. Soc., Perkin Trans. 1 1980, 2380.
- ↑ Carboni, S.; Da Settimo, A.; Bertini, D.; Ferrarini, P. L.; Livi, O.; Tonetti, I. Farmaco, Ed. Sci. 1975, 30, 237.
- ↑ Trost, B. M.; Vaultier, M.; Santiago, M. L. J. Am. Chem. Soc. 1980, 102, 7929.
- ↑ Aubé, J.; Milligan, G. L. J. Am. Chem. Soc. 1991, 113, 8965.
- ↑ Gracias, V.; Milligan, G. L.; Aubé, J. J. Am. Chem. Soc. 1995, 117, 8047.
- ↑ Pearson, W. H.; Fang, W.-k. J. Org. Chem. 2000, 65, 7158.
- ↑ Pearson, W. H. J. Heterocycl. Chem. 1996, 33, 1489.
- ↑ Reddy, P. G.; Varghese, B.; Baskaran, S. Org. Lett. 2003, 5, 583.
- ↑ Georg, G. I.; Guan, X.; Kant, J. Tetrahedron Lett. 1988, 29, 403.
- ↑ Le Dreau, M. A.; Desmaele, D.; Dumas, F.; d'Angelo, J. J. Org. Chem. 1993, 58, 2933.
- ↑ Gawley, R. E. Org. React. 1988, 35, 1. doi: (10.1002/0471264180.or035.01)
- ↑ Oliveros-Desherces, E.; Rivière, M.; Parello, J.; Lattes, A. Tetrahedron Lett. 1975, 851.
- ↑ Lebel, H.; Leogane, O. Org. Lett. 2005, 7, 4107.
- ↑ Peet, N. P.; Weintraub, P. M. Chem. Eng. News 1994, 72, 4.
- ↑ Viaud, M. C.; Rollin, P. Synthesis 1990, 130.
- ↑ Ito, M.; Koyakumaru, K.-i.; Ohta, T.; Takaya, H. Synthesis 1995, 1995, 376.
- ↑ Merour, J. Y.; Cossais, F.; Piroelle, S.; Mazeas, D. J. Heterocycl. Chem. 1994, 31, 87.