
6.4: Rotational Partition Function
- Page ID
- 285779
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Rigid Rotor Assumption and Rotamers
Separation of the rotational partition function from the partition functions of other degrees of freedom does not only require consideration of nuclear spin states, but also the assumption that the moment of inertia is the same for all rotational states. This is generally true if the molecule behaves as a rigid rotor. For small molecules consisting of only a few atoms, this is often a good approximation. Larger molecules feature internal rotations, where a group of atoms rotates with respect to the rest of the molecule. In general, internal rotations are torsions about rotatable bonds, which are often not freely rotatable. The torsion potential has several minima and these minima are separated by energy barriers with heights that are larger, but not much larger than \(k_\mathrm{B} T\). If we denote the number of potential minima for the \(i^\mathrm{th}\) rotatable bond with \(n_{\mathrm{min},i}\), the total number of rotameric states, short rotamers is
\[n_\mathrm{rot} = \prod_i n_{\mathrm{min},i} \ .\]
Each rotamer has it’s own moment of inertia and, hence, its own set of states with respect to total rotation of the molecule. Because the energy scales of internal and total rotations are not well separated and because in larger molecules some vibrational modes may also have energies in this range, the partition function is not usually separable for large molecules. In such cases, insight into statistical thermodynamics can be best obtained by MD simulations. In the following, we consider small molecules that can be assumed to behave as a rigid rotor. We first consider diatomic molecules, where it certainly applies on the level of precision that we seek here.
The energy levels of a rigid rotor of a diatomic molecule are quantized by the rotational quantum number \(J\) and given by
\[\epsilon_J = J(J+1) \frac{h^2}{8\pi^2 I} = h c B J (J+1) \ ,\]
where
\[I = \mu r^2\]
is the moment of inertia with the reduced mass \(\mu\) and
\[B = \frac{h}{8 \pi^2 I c}\]
is the rotational constant. After introducing the characteristic rotational temperature,
\[\Theta_\mathrm{rot} = \frac{h^2}{8 \pi^2 I k_\mathrm{B}} = \frac{h c B}{k_\mathrm{B}}\]
we have
\[\epsilon_J = J(J+1) k_\mathrm{B} \Theta_\mathrm{rot} \ .\]
As already mentioned, each rotational level has a degeneracy \(g_J = 2 J + 1\). If all nuclei in the molecules are distinguishable (magnetically not equivalent), there is no correlation with nuclear spin states. In that case we have
\[Z_\mathrm{rot} = \sum \left( 2 J + 1 \right) e^{-J(J+1)\Theta_\mathrm{rot}/T} \ . \label{eq:rot_sum}\]
For sufficiently high temperatures and a sufficiently large moment of inertia, the density of states is sufficiently large for replacing the sum by an integral,
\[Z_\mathrm{rot} \approx \int_0^\infty \left( 2 J + 1 \right) e^{-J(J+1)\Theta_\mathrm{rot}/T} \mathrm{d}J = \frac{T}{\Theta_\mathrm{rot}} \ . \label{eq:continuum}\]
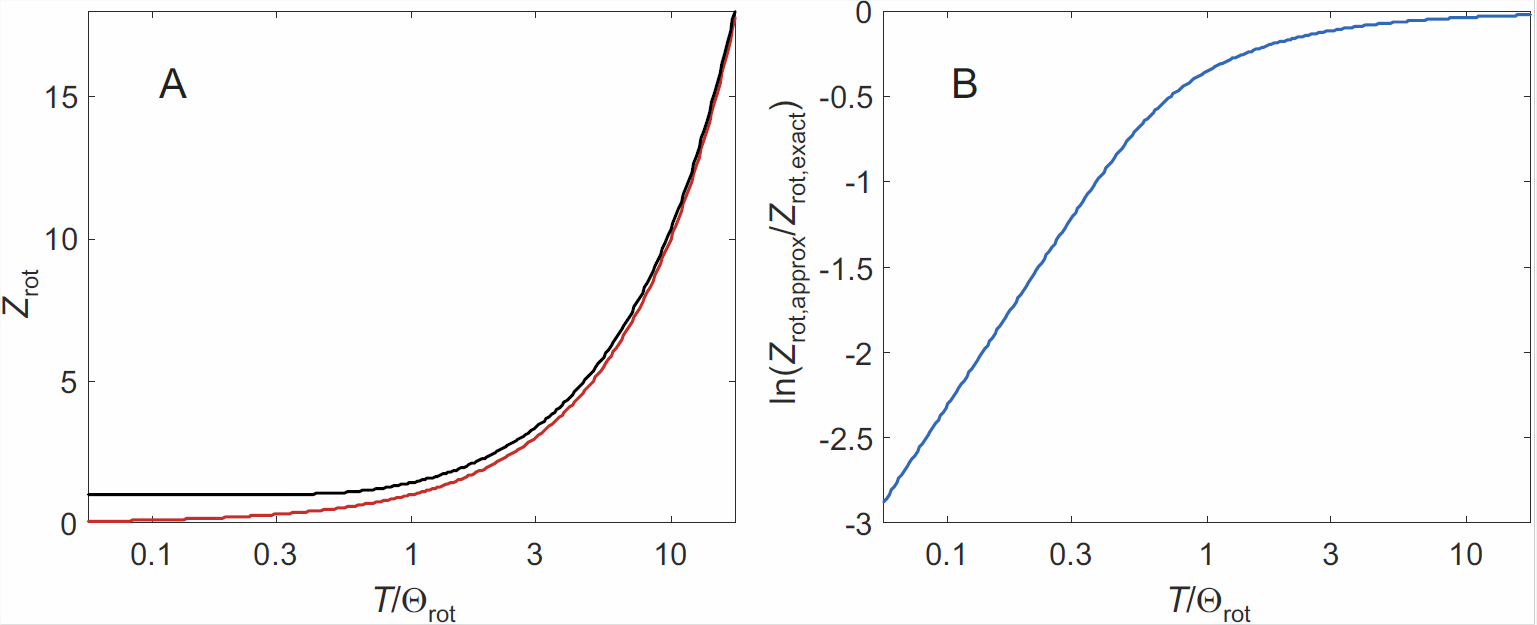
Deviations between the partition functions computed by Equation \ref{eq:rot_sum}) and Equation \ref{eq:continuum}) are visualized in Figure \(\PageIndex{1}\). As state functions depend on \(\ln Z\), the continuum approximation is good for \(T/\Theta_\mathrm{rot} \gg 1\), which applies to all gases, except at low temperatures for those that contain hydrogen. At ambient temperature it can be used in general, except for a further correction that we need to make because of symmetry considerations.
Accessible States and Symmetry
Even if all nuclei are magnetically inequivalent and, hence, distinguishable, rotational states may be not. For heteronuclear diatomic molecules they are, but for homonuclear diatomic molecules, they are not. To see this, we consider a symmetrical linear molecule that rotates by 180\(^\circ\) about an axis perpendicular to the bond axis and centered in the middle of the bond. This rotation produces a configuration that is indistinguishable from the original configuration. In other words, the nuclear wavefunction is symmetric with respect to this rotation. For a homonuclear diatomic molecule, half of the rotational states are symmetric and half are antisymmetric. For nuclei that are bosons, such as \(\ce{^{16}O}\) in dioxygen, only the former states are allowed, for nuclei that are fermions, only the latter are allowed. Hence, we need to divide the partition function, which counts accessible states, by two. In this example, we have deliberately chosen a case with only one nuclear spin state. If nuclear spin states with different symmetry exist, all rotational states are accessible, but correlated to the nuclear spin states, as we have seen before for dihydrogen. In the following we consider the situation with only one nuclear spin state or for a fixed nuclear spin state.
| Group | \(\sigma\) | Group | \(\sigma\) | Group | \(\sigma\) | Group | \(\sigma\) |
|---|---|---|---|---|---|---|---|
| \(C_1\), \(C_i\), \(C_s\), \(C_{\infty v}\) | 1 | \(D_{\infty h}\) | 2 | \(T\), \(T_d\) | 12 | \(O_h\) | 24 |
| \(C_n\), \(C_{nv}\), \(C_{nh}\) | \(n\) | \(D_n\), \(D_{nh}\), \(D_{nd}\) | \(2 n\) | \(S_n\) | \(n/2\) | \(I_h\) | 60 |
Although we still did not discuss other complications for multi-atomic molecules, we generalize this concept of symmetry-accessible states by introducing a symmetry number \(\sigma\). In general, \(\sigma\) is the number of distinct orientations of a rigid molecule that are distinguished only by interchange of identical atoms. For an NH\(_3\) molecule, rotation about the \(C_3\) axis by 120\(^\circ\) generates one such orientation from the other. No other rotation axis exists. Hence, \(\sigma = 3\) for NH\(_3\). In general, the symmetry number can be obtained from the molecule’s point group , as shown in Table \(\PageIndex{1}\).
With the symmetry number, the continuum approximation (Equation \ref{eq:continuum} becomes
\[Z_\mathrm{rot} = \frac{T}{\sigma \Theta_\mathrm{rot}} \ , \label{eq:Z_rot}\]
where we still assume that symmetry is sufficiently high for assigning a single characteristic temperature.
We further find
\[\ln Z_\mathrm{rot} = \ln T + \ln \frac{1}{\sigma \Theta_\mathrm{rot}} \ , \label{eq:ln_Z_rot}\]
and with this
\[\begin{align} U_\mathrm{rot} = N_\mathrm{Av} k_\mathrm{B} T^2 \left( \frac{\partial \ln Z_\mathrm{rot}}{\partial T} \right)_V \label{eq:u_rot0} \\ = R T^2 \frac{\partial}{\partial T} \ln T \label{eq:u_rot1} \\ = R T \ \textrm{(linear)} \ .\end{align}\]
On going from Equation \ref{eq:u_rot0} to \ref{eq:u_rot1} we could drop the second term on the right-hand side of Equation \ref{eq:ln_Z_rot}, as this term does not depend on temperature. This is a general principle: Constant factors in the partition function do not contribute to internal energy. The result can be generalized to multi-atomic linear molecules that also have two rotational degrees of freedom. This result is expected from the equipartition theorem, as each degree of freedom should contribute a term \(k_\mathrm{B} T/2\) to the molecular or a term \(R T/2\) to the molar internal energy. However, if we refrain from the continuum approximation in Equation \ref{eq:continuum}) and numerically evaluate Equation \ref{eq:rot_sum}) instead, we find a lower contribution for temperatures lower than or comparable to \(\Theta_\mathrm{rot}\). This is also a general principle: Contributions of modes to internal energy and, by inference, to heat capacity, are fully realized only at temperatures much higher than their characteristic temperature and are negligible at temperatures much lower than their characteristic temperature.20
For the rotation contribution to molar heat capacity at constant volume of a linear molecule we obtain
\[C_{\mathrm{rot},V} = \frac{\partial}{\partial T} U_\mathrm{rot} = R \ .\]
A non-linear multi-atomic molecule has, in general, three independent rotational moments of inertia corresponding to three pairwise orthogonal directions \(a, b, c\). With
\[\Theta_{\mathrm{rot},\alpha} = \frac{h^2}{8 \pi^2 I_\alpha k_\mathrm{B}} \ \left( \alpha = a \ , b \ , c \right)\]
one finds for the partition function
\[Z_\mathrm{rot} = \frac{\sqrt{\pi}}{\sigma} \left( \frac{T^3}{\Theta_{\mathrm{rot},a} \Theta_{\mathrm{rot},b} \Theta_{\mathrm{rot},c}} \right)^{1/2} \ .\]
For spherical-top molecules, all three moments of inertia are equal, \(I_a = I_b = I_c\), and hence all three characteristic temperatures are equal. For symmetric-top molecules, \(I_a = I_b \neq I_c\).
The general equations for \(U_\mathrm{rot}\) and \(C_{\mathrm{rot},V}\) at sufficiently high temperature \(T \gg \Theta_\mathrm{rot}\) are
\[\begin{align} U_\mathrm{rot} & = \frac{d}{2} R T \\ C_{\mathrm{rot},V} & = \frac{d}{2} R \ ,\end{align}\]
where \(d = 1\) for a free internal rotation (e.g., about a C\(\equiv\)C bond), \(d=2\) for linear, and \(d=3\) for non-linear molecules. We note that the contribution of a free internal rotation needs to be added to the contribution from total rotation of the molecule.
The expressions for the rotational contributions to molar entropy are a bit lengthy and do not provide additional physical insight. They can be easily derived from the appropriate expressions for the rotational partition function and internal energy using Equation \ref{eq:s_from_z}). We note, however, that the contribution from the \(u/T\) term in the entropy expression is \(d R/2\) and the contribution from \(\ln Z\) is positive. Hence, at temperatures where the continuum approximation is valid, the rotational contribution to entropy is larger than \(d R/2\).