
3.35: Semi-empirical Molecular Orbital Calculation on XeF2
- Page ID
- 154842

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The bonding in XeF2 can be interpreted in terms the three-center four-electron bond. In linear XeF2 the molecular orbital can be considered to be a linear combination of the fluorine 2p orbitals and the central xenon 5p.
\[ \Psi = C_{F1} \Psi_{F1} + C_{Xe} \Psi_{Xe} + C_{F2} \Psi_{F2} \nonumber \]
Minimization of the variational integral
\[ E = \frac{ \int \left( C_{F1} \Psi_{F1} + C_{Xe} \Psi_{Xe} + C_{F2} \Psi_{F2} \right) H \left( C_{F1} \Psi_{F1} + C_{Xe} \Psi_{Xe} + C_{F2} \Psi_{F2} \right) d \tau}{ \int \left( C_{F1} \Psi_{F1} + C_{Xe} \Psi_{Xe} + C_{F2} \Psi_{F2} \right)^2 d \tau} \nonumber \]
yields the following 3 x 3 Huckel matrix
\[ H = \begin{pmatrix} \alpha_F & \beta & 0 \\ \beta & \alpha_{Xe} & \beta \\ 0 & \beta & \alpha_F \end{pmatrix} \nonumber \]
All overlap integrals are zero. The Coulomb integrals are parameterized as the negative of the valence orbital ionization energies (-12.13 eV for the Xe 5p orbital, -17.42 eV for the F 2p). The non-zero resonance integral is given a value of -2.0 eV.
\[ \begin{matrix} \alpha_{F1} = \alpha_F = \int \Psi_{F1} H \Psi_{F1} d \tau = -17.42 & \alpha_{F2} = \alpha_F = \int \Psi_{F2} H \Psi_{F2} d \tau = -17.42 \\ \alpha_{Xe} = \int \Psi_{Xe} H \Psi_{Xe} d \tau = -12.13 & \beta= \int \Psi_{Xe} H \Psi_F d \tau = -2 \end{matrix} \nonumber \]
Mathcad can now be used to find the eigenvalues and eigenvectors of H. First we must give it the values for all of the parameters.
\[ \begin{matrix} \alpha_F = -17.42 & \alpha_{Xe} = -12.13 & \beta = -2.00 \end{matrix} \nonumber \]
Define the variational matrix shown on the left side of equation (6):
\[ H = \begin{pmatrix} \alpha_F & \beta & 0 \\ \beta & \alpha_{Xe} & \beta \\ 0 & \beta & \alpha_F \end{pmatrix} \nonumber \]
Find the eigenvalues: \( \begin{matrix} \text{E = sort(eigenvals(H))} & E_0 = -18.647 & E_1 = -17.42 & E_2 = -10.903 \end{matrix}\)
Now find the eigenvectors:
\[ \begin{matrix} \text{The bonding MO is:} & \Psi_b = \text{eigenvec} \left( H,~E_0 \right) & \Psi_b = \begin{pmatrix} 0.649 \\ 0.389 \\ 0.649 \end{pmatrix} & \overrightarrow{ \left( \Psi_b \right)^2} = \begin{pmatrix} 0.421 \\ 0.158 \\ 0.421 \end{pmatrix} \\ \text{The non-bonding MO is:} & \Psi_{nb} = \text{eigenvec} \left( H,~E_1 \right) & \Psi_{nb} = \begin{pmatrix} -0.707 \\ 0 \\ 0.707 \end{pmatrix} & \overrightarrow{ \left( \Psi_{nb} \right)^2} = \begin{pmatrix} 0.5 \\ 0 \\ 0.5 \end{pmatrix} \\ \text{The anti-bonding MO is:} & \Psi_a = \text{eigenvec} \left( H,~E_2 \right) & \Psi_b = \begin{pmatrix} -0.282 \\ 0.917 \\ -0.282 \end{pmatrix} & \overrightarrow{ \left( \Psi_a \right)^2} = \begin{pmatrix} 0.079 \\ 0.842 \\ 0.079 \end{pmatrix} \end{matrix} \nonumber \]
The molecular orbital diagram for this system is shown below.
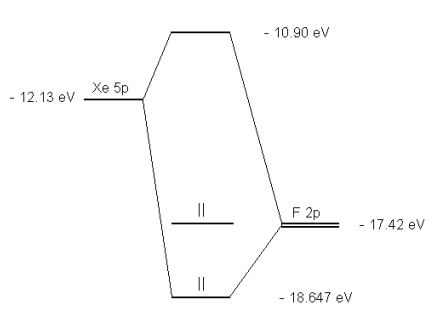
Is the molecule stable? The diagram shows two electrons each in the bonding and non-bonding orbitals for a total energy of -72.14 eV. The energy of the isolated atoms is 2(-12.13 eV) + 2(-17.42 eV) = - 59.1 eV. Thus, the molecule is more stable than the isolated atoms according to this crude semi-empirical model.
The partial charges are calculated next. The fluorine atoms have a kernel charge of +7. They get credit for 6 non-bonding atomic electrons, 42.1% of the two electrons in the bonding MO, and 50% of the two electrons in the non-bonding MO. This gives a partial charge of (7 - 6 - 2(.421) - 2(.50)) -0.842.
The xenon atom has a kernel charge of +8. It gets credit for 6 non-bonding atomic electrons, 15.8% of the two electrons in the bonding MO, and no credit for the two electrons in non-bonding MO. This yields a partial charge of (8 - 6 - 2(.158)) +1.684. This value is reasonable because xenon is bonded to the most electronegative element in the periodic table.