
14.4: Electronic Spectroscopy
- Page ID
- 41397
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)This page explains what happens when organic compounds absorb UV or visible light, and why the wavelength of light absorbed varies from compound to compound.
Molecular Absorption of Light
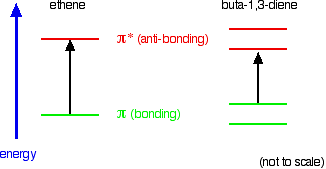
When we were talking about the various sorts of molecular orbitals present in organic compounds earlier, you will have come across this diagram showing their relative energies:

When light passes through the compound, energy from the light is used to promote an electron from a bonding or non-bonding orbital into one of the empty anti-bonding orbitals. The possible electron jumps that light might cause are:

In each possible case, an electron is excited from a full orbital into an empty anti-bonding orbital. Each jump takes energy from the light, and a big jump obviously needs more energy than a small one. Each wavelength of light has a particular energy associated with it. If that particular amount of energy is just right for making one of these energy jumps, then that wavelength will be absorbed - its energy will have been used in promoting an electron.
An absorption spectrometer works in a range from about 200 nm (in the near ultra-violet) to about 800 nm (in the very near infra-red). Only a limited number of the possible electron jumps absorb light in that region. Look again at the possible jumps. This time, the important jumps are shown in black, and a less important one in grey. The grey dotted arrows show jumps which absorb light outside the region of the spectrum we are working in.
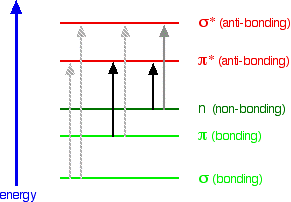
Remember that bigger jumps need more energy and so absorb light with a shorter wavelength. The jumps shown with grey dotted arrows absorb UV light of wavelength less that 200 nm. The important jumps are:
- from \(\pi\) bonding orbitals to \(\pi\) anti-bonding orbitals;
- from non-bonding orbitals to \(\pi\) anti-bonding orbitals;
- from non-bonding orbitals to sigma anti-bonding orbitals.
That means that in order to absorb light in the region from 200 - 800 nm (which is where the spectra are measured), the molecule must contain either \(\pi\) bonds or atoms with non-bonding orbitals. Remember that a non-bonding orbital is a lone pair on, say, oxygen, nitrogen or a halogen. Groups in a molecule which absorb light are known as chromophores.
What does an absorption spectrum look like
The diagram below shows a simple UV-visible absorption spectrum for buta-1,3-diene - a molecule we will talk more about later. Absorbance (on the vertical axis) is just a measure of the amount of light absorbed. The higher the value, the more of a particular wavelength is being absorbed.
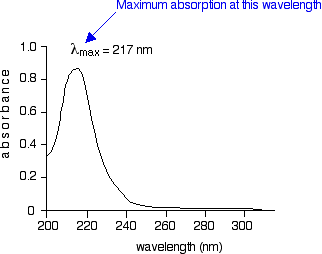
You will see that absorption peaks at a value of 217 nm. This is in the ultra-violet and so there would be no visible sign of any light being absorbed - buta-1,3-diene is colorless. In buta-1,3-diene, CH2=CH-CH=CH2, there are no non-bonding electrons. That means that the only electron jumps taking place (within the range that the spectrometer can measure) are from \(\pi\) bonding to \(\pi\) anti-bonding orbitals.
A chromophore such as the carbon-oxygen double bond in ethanal, for example, obviously has \(\pi\) electrons as a part of the double bond, but also has lone pairs on the oxygen atom. That means that both of the important absorptions from the last energy diagram are possible. You can get an electron excited from a \(\pi\) bonding to a \(\pi\) anti-bonding orbital, or you can get one excited from an oxygen lone pair (a non-bonding orbital) into a \(\pi\) anti-bonding orbital.
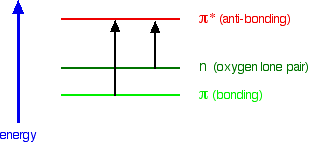
The non-bonding orbital has a higher energy than a \(\pi\) bonding orbital. That means that the jump from an oxygen lone pair into a \(\pi\) anti-bonding orbital needs less energy. That means it absorbs light of a lower frequency and therefore a higher wavelength. For example, ethanal can therefore absorb light of two different wavelengths:
- the \(\pi\) bonding to \(\pi\) anti-bonding absorption peaks at 180 nm. These \(n \rightarrow \pi^*\) transitions involve moving an electron from a nonbonding electron pair to a antibonding \*pi^*\) orbital. They tend to have molar absorbtivities less than 2000
- the non-bonding to \(\pi\) anti-bonding absorption peaks at 290 nm. These \(\pi \rightarrow \pi^*\) transitions involve moving an electron from a bonding \(\pi*\) orbital to an antibonding \(\pi^*\) orbital. They tend to have molar absorptivities on the order of 10,000.
Both of these absorptions are in the ultra-violet, but most spectrometers will not pick up the one at 180 nm because they work in the range from 200 - 800 nm.
Conjugation and Delocalization
Consider these three molecules:
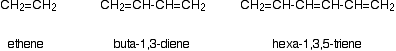
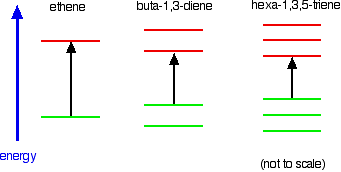
Ethene contains a simple isolated carbon-carbon double bond, but the other two have conjugated double bonds. In these cases, there is delocalization of the \(\pi\) bonding orbitals over the whole molecule. Now look at the wavelengths of the light which each of these molecules absorbs.
| molecule | wavelength of maximum absorption (nm) |
|---|---|
| ethene | 171 |
| buta-1,3-diene | 217 |
| hexa-1,3,5-triene | 258 |
All of the molecules give similar UV-visible absorption spectra - the only difference being that the absorptions move to longer and longer wavelengths as the amount of delocalization in the molecule increases. Why is this? You can actually work out what must be happening.
- The maximum absorption is moving to longer wavelengths as the amount of delocalization increases.
- Therefore maximum absorption is moving to shorter frequencies as the amount of delocalization increases.
- Therefore absorption needs less energy as the amount of delocalization increases.
- Therefore there must be less energy gap between the bonding and anti-bonding orbitals as the amount of delocalization increases.
. . . and that's what is happening.
Compare ethene with buta-1,3-diene. In ethene, there is one \(\pi\) bonding orbital and one \(\pi\) anti-bonding orbital. In buta-1,3-diene, there are two \(\pi\) bonding orbitals and two \(\pi\) anti-bonding orbitals. This is all discussed in detail on the introductory page that you should have read.
The highest occupied molecular orbital is often referred to as the HOMO - in these cases, it is a \(\pi\) bonding orbital. The lowest unoccupied molecular orbital (the LUMO) is a \(\pi\) anti-bonding orbital. Notice that the gap between these has fallen. It takes less energy to excite an electron in the buta-1,3-diene case than with ethene. In the hexa-1,3,5-triene case, it is less still.
If you extend this to compounds with really massive delocalization, the wavelength absorbed will eventually be high enough to be in the visible region of the spectrum, and the compound will then be seen as colored. A good example of this is the orange plant pigment, beta-carotene - present in carrots, for example.
Why is beta-carotene orange?
Beta-carotene has the sort of delocalization that we've just been looking at, but on a much greater scale with 11 carbon-carbon double bonds conjugated together. The diagram shows the structure of beta-carotene with the alternating double and single bonds shown in red.
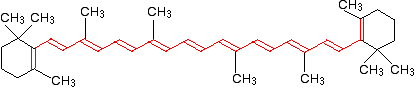
The more delocalization there is, the smaller the gap between the highest energy \(\pi\) bonding orbital and the lowest energy \(\pi\) anti-bonding orbital. To promote an electron therefore takes less energy in beta-carotene than in the cases we've looked at so far - because the gap between the levels is less. Remember that less energy means a lower frequency of light gets absorbed - and that's equivalent to a longer wavelength. Beta-carotene absorbs throughout the ultra-violet region into the violet - but particularly strongly in the visible region between about 400 and 500 nm with a peak about 470 nm. The wavelengths associated with the various colors are approximately:
| color region | wavelength (nm) |
|---|---|
| violet | 380 - 435 |
| blue | 435 - 500 |
| cyan | 500 - 520 |
| green | 520 - 565 |
| yellow | 565 - 590 |
| orange | 590 - 625 |
| red | 625 - 740 |
So if the absorption is strongest in the violet to cyan region, what color will you actually see? It is tempting to think that you can work it out from the colors that are left - and in this particular case, you wouldn't be far wrong. Unfortunately, it is not as simple as that! Sometimes what you actually see is quite unexpected. Mixing different wavelengths of light does not give you the same result as mixing paints or other pigments. You can, however, sometimes get some estimate of the color you would see using the idea of complementary colors. If you arrange some colors in a circle, you get a "color wheel". The diagram shows one possible version of this. An internet search will throw up many different versions!
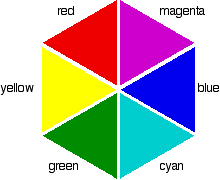
colors directly opposite each other on the color wheel are said to be complementary colors. Blue and yellow are complementary colors; red and cyan are complementary; and so are green and magenta. Mixing together two complementary colors of light will give you white light. What this all means is that if a particular color is absorbed from white light, what your eye detects by mixing up all the other wavelengths of light is its complementary color. In the beta-carotene case, the situation is more confused because you are absorbing such a range of wavelengths. However, if you think of the peak absorption running from the blue into the cyan, it would be reasonable to think of the color you would see as being opposite that where yellow runs into red - in other words, orange.
Franck-Condon: Electronic and Vibrational Coupling
So far, we have come across one big rule of photon absorbance. To be absorbed, a photon's energy has to match an energy difference within the compound that is absorbing it.
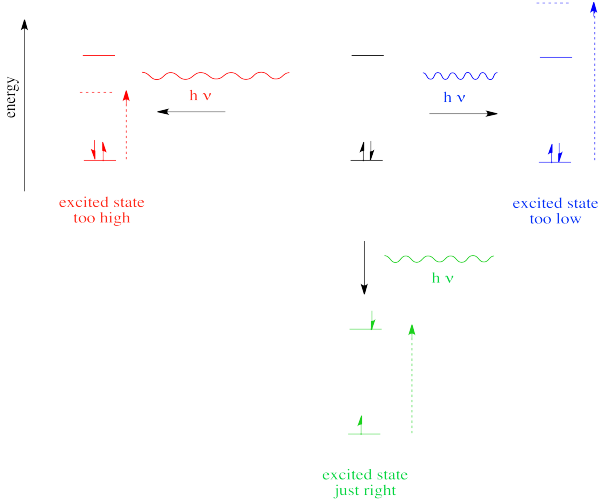
In the case of visible or ultraviolet light, the energy of a photon is roughly in the region that would be appropriate to promote an electron to a higher energy level. Different wavelengths would be able to promote different electrons, depending on the energy difference between an occupied electronic energy level and an unoccupied one. Other types of electromagnetic radiation would not be able to promote an electron, but they would be coupled to other events. For example, absorption of infrared light is tied to vibrational energy levels. Microwave radiation is tied to rotational energy levels in molecules. Thus, one reason a photon may or may not be absorbed has to do with whether its energy corresponds to the available energy differences within the molecule or ion that it encounters.
Photons face other limitations. One of these is a moderate variation on our main rule. It is called the Frank Condon Principle. According to this idea, when an electron is excited from its normal position, the ground state, to a higher energy level, the optimal positions of atoms in the molecule may need to shift. Because electronic motion is much faster than nuclear motion, however, any shifting of atoms needed to optimize positions as they should be in the excited state will have to wait until after the electron gets excited. In that case, when the electron lands and the atoms aren't yet in their lowest energy positions for the excited state, the molecule will find itself in an excited vibrational state as well as an excited electronic state.

That means the required energy for excitation does not just correspond to the difference in electronic energy levels; it is fine-tuned to reach a vibrational energy level, which is quantized as well.
- The Franck Condon Principle states that electronic transitions are vertical.
- A vertical transition is one in which non of the nuclei move while the electron journeys from one state to another.
- A vertical transition may begin in a vibrational ground state of an electronic ground state and end in a vibrational excited state of an electronic excited state.