
B. What is Nucleophilic Substitution?
- Page ID
- 3777

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Halogenoalkanes (also known as haloalkanes or alkyl halides) are compounds containing a halogen atom (fluorine, chlorine, bromine or iodine) joined to one or more carbon atoms in a chain. The interesting thing about these compounds is the carbon-halogen bond, and all the nucleophilic substitution reactions of the halogenoalkanes involve breaking that bond. With the exception of iodine, all of the halogens are more electronegative than carbon.
| C | 2.5 |
| F | 4.0 |
| Cl | 3.0 |
| Br | 2.8 |
| I | 2.5 |
That means that the electron pair in the carbon-halogen bond will be dragged towards the halogen end, leaving the halogen slightly negative (![]() -) and the carbon slightly positive (
-) and the carbon slightly positive (![]() +) - except in the carbon-iodine case. Although the carbon-iodine bond doesn't have a permanent dipole, the bond is very easily polarized by anything approaching it. Imagine a negative ion approaching the bond from the far side of the carbon atom:
+) - except in the carbon-iodine case. Although the carbon-iodine bond doesn't have a permanent dipole, the bond is very easily polarized by anything approaching it. Imagine a negative ion approaching the bond from the far side of the carbon atom:
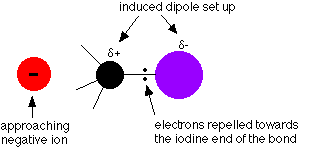
The fairly small polarity of the carbon-bromine bond will be increased by the same effect.
The strengths of the carbon-halogen bonds
In all of these nucleophilic substitution reactions, the carbon-halogen bond has to be broken at some point during the reaction. In general, the harder it is to break, the slower the reaction will be. Look at the strengths of various bonds (all values in kJ mol-1).
| C-H | 413 |
| C-F | 467 |
| C-Cl | 346 |
| C-Br | 290 |
| C-I | 228 |
The carbon-fluorine bond is very strong (stronger than C-H) and isn't easily broken. It doesn't matter that the carbon-fluorine bond has the greatest polarity - the strength of the bond is much more important in determining its reactivity. You might therefore expect fluoroalkanes to be very unreactive - and they are! We shall simply ignore them from now on. In the other halogenoalkanes, the bonds get weaker as you go from chlorine to bromine to iodine. That means that chloroalkanes react most slowly, bromoalkanes react faster, and iodoalkanes react faster still.
Rates of reaction: RCl < RBr < RI
Where "<" is read as "is less than" - or, in this instance, "is slower than", and R represents any alkyl group.
Nucleophilic substitution in primary halogenoalkanes
You will need to know about this if your syllabus talks about "primary halogenoalkanes" or about SN2 reactions. If the syllabus is vague, check recent exam papers and mark schemes, and compare them against what follows.
A nucleophile is a species (an ion or a molecule) which is strongly attracted to a region of positive charge in something else. Nucleophiles are either fully negative ions, or else have a strongly 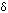 - charge somewhere on a molecule. Common nucleophiles are hydroxide ions, cyanide ions, water and ammonia.
- charge somewhere on a molecule. Common nucleophiles are hydroxide ions, cyanide ions, water and ammonia.
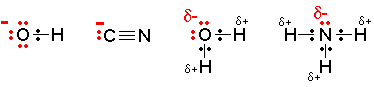
Notice that each of these contains at least one lone pair of electrons, either on an atom carrying a full negative charge, or on a very electronegative atom carrying a substantial ![]() - charge.
- charge.
The nucleophilic substitution reaction - an SN2 reaction
We'll talk this mechanism through using an ion as a nucleophile, because it's slightly easier. The water and ammonia mechanisms involve an extra step which you can read about on the pages describing those particular mechanisms. We'll take bromoethane as a typical primary halogenoalkane. The bromoethane has a polar bond between the carbon and the bromine.
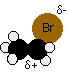
We'll look at its reaction with a general purpose nucleophilic ion which we'll call Nu-. This will have at least one lone pair of electrons. Nu- could, for example, be OH- or CN-.
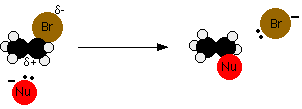
The lone pair on the Nu- ion will be strongly attracted to the ![]() + carbon, and will move towards it, beginning to make a co-ordinate (dative covalent) bond. In the process the electrons in the C-Br bond will be pushed even closer towards the bromine, making it increasingly negative.
+ carbon, and will move towards it, beginning to make a co-ordinate (dative covalent) bond. In the process the electrons in the C-Br bond will be pushed even closer towards the bromine, making it increasingly negative.
The movement goes on until the -Nu is firmly attached to the carbon, and the bromine has been expelled as a Br- ion.
The Nu- ion approaches the \(\delta^+\) carbon from the side away from the bromine atom. The large bromine atom hinders attack from its side and, being \(\delta^-\), would repel the incoming Nu- anyway. This attack from the back is important if you need to understand why tertiary halogenoalkanes have a different mechanism. We'll discuss this later on this page.
There is obviously a point in which the Nu- is half attached to the carbon, and the C-Br bond is half way to being broken. This is called a transition state. It isn't an intermediate. You can't isolate it - even for a very short time. It's just the mid-point of a smooth attack by one group and the departure of another.
Nucleophile effects
A major factor affecting the SN2 reaction is strength of the nucleophile. Recall that a nucleophile is a molecule which is attracted to positive charge. We can describe several general trends which determine the strength of a nucleophile:
- A molecule which has lost a proton and is negatively charged (base) is stronger than the neutral, protonated version of the same molecule (conjugate acid).
- Nucleophilicity of the attacking atom decreases from left to right in the periodic table because atoms because atoms are more electronegative (hold electrons more closely) going from left to right.
- Nucleophilicity of the attacking atom increases down the periodic table as electronegativity decreases and polarizibity increases. Polarizibility refers to the ability of larger atoms lower in the periodic table to share electrons more easily because their electrons are more loosely held.
In addition to the strength of the nucleophile, the structure of the nucleophile also influences the capacity of an SN2 reaction to proceed. As stated many times previously, steric effects are the most important factor in determining whether an SN2 reaction can proceed. We have discussed these effects with regard to the substrate, but they also apply to the nucleophile. A small, unbranched nucleophile will be more effective in an SN2 reaction than a large, branched nucleophile.
Substrate effects
Based on the factors described above, we can outline several trends that affect the likelihood of an SN2 reaction occurring. As described above, the most important factor in deciding whether an SN2 reaction occurs is steric effects. When we describe the nature of the molecule being attacked, called the substrate, we can look at how many alkyl substituents are present on the molecule. For example, the molecule may contain a carbon atom attached to a leaving group and three hydrogen atoms. This would constitute a methyl carbon atom, in analogy to methane. Given that such a molecule has almost no steric hindrance (substituents blocking access to the carbon atom), an SN2 reaction will be highly favored in a molecule with a methyl carbon.
We call a carbon atom with one alkyl substituent a primary carbon atom. SN2 reactions will proceed easily with a primary carbon atom, though not as fast as with a methyl carbon. If there are two alkyl substituents, we call the carbon atom secondary. It is possible for an SN2 reaction to proceed at a secondary carbon atom, but it is not as favorable as in the methyl or primary cases. Finally, if there are three alkyl substituents attached to the carbon atom, we have the case of a tertiary carbon atom. In this case, there is far too much steric hindrance, and the SN2 reaction cannot proceed. The principle that increasing substitution leads to decreasing reactivity is outlined in the following table:
| Relative rate of SN2 reaction | ||||||
| Methyl | > | Primary | > | Secondary | > | Tertiary |
The degree of substitution of the molecule undergoing attack is not the only factor that influences the rate of an SN2 reaction. Recall that steric effects (the likelihood that attack by the nucleophile will be blocked) are of paramount importance in determining whether an SN2 reaction will take place. Degree of substitution is one factor in determining steric effects, but it is not the only one. We also need to consider the bulkiness of the substituents.
How to write the mechanism
The simplest way is:
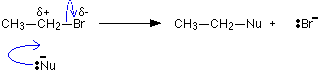
Technically, this is known as an SN2 reaction. S stands for substitution, N for nucleophilic, and the 2 is because the initial stage of the reaction involves two species - the bromoethane and the Nu- ion. If your syllabus doesn't refer to SN2 reactions by name, you can just call it nucleophilic substitution.
Some examiners like you to show the transition state in the mechanism, in which case you need to write it in a bit more detail - showing how everything is arranged in space.
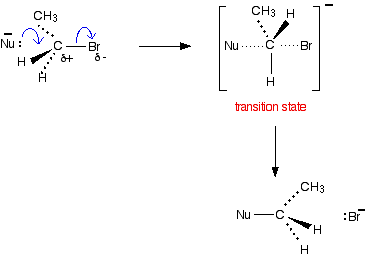
Be very careful when you draw the transition state to make a clear difference between the dotted lines showing the half-made and half-broken bonds, and those showing the bonds going back into the paper. Notice that the molecule has been inverted during the reaction - rather like an umbrella being blown inside-out.
Nucleophilic substitution in tertiary halogenoalkanes
Remember that a tertiary halogenoalkane has three alkyl groups attached to the carbon with the halogen on it. These alkyl groups can be the same or different, but in this section, we shall just consider a simple one, (CH3)3CBr - 2-bromo-2-methylpropane.
The nucleophilic substitution reaction - an SN1 reaction
Once again, we'll talk this mechanism through using an ion as a nucleophile, because it's slightly easier, and again we'll look at the reaction of a general purpose nucleophilic ion which we'll call Nu-. This will have at least one lone pair of electrons.
Why is a different mechanism necessary?
You will remember that when a nucleophile attacks a primary halogenoalkane, it approaches the ![]() + carbon atom from the side away from the halogen atom. With a tertiary halogenoalkane, this is impossible. The back of the molecule is completely cluttered with CH3 groups.
+ carbon atom from the side away from the halogen atom. With a tertiary halogenoalkane, this is impossible. The back of the molecule is completely cluttered with CH3 groups.
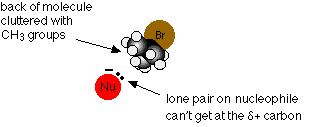
Since any other approach is prevented by the bromine atom, the reaction has to go by an alternative mechanism.
The alternative mechanism
The reaction happens in two stages. In the first, a small proportion of the halogenoalkane ionises to give a carbocation and a bromide ion.
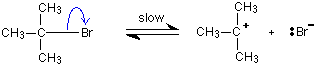
This reaction is possible because tertiary carbocations are relatively stable compared with secondary or primary ones. Even so, the reaction is slow. Once the carbocation is formed, however, it would react immediately it came into contact with a nucleophile like Nu-. The lone pair on the nucleophile is strongly attracted towards the positive carbon, and moves towards it to create a new bond.
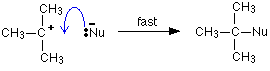
How fast the reaction happens is going to be governed by how fast the halogenoalkane ionises. Because this initial slow step only involves one species, the mechanism is described as SN1 - substitution, nucleophilic, one species taking part in the initial slow step.
Why don't primary halogenoalkanes use the SN1 mechanism?
If a primary halogenoalkane did use this mechanism, the first step would be, for example:
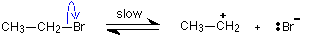
A primary carbocation would be formed, and this is much more energetically unstable than the tertiary one formed from tertiary halogenoalkanes - and therefore much more difficult to produce. This instability means that there will be a very high activation energy for the reaction involving a primary halogenoalkane. The activation energy is much less if it undergoes an SN2 reaction - and so that's what it does instead.
Nucleophilic substitution in secondary halogenoalkanes
There isn't anything new in this. Secondary halogenoalkanes will use both mechanisms - some molecules will react using the SN2 mechanism and others the SN1.
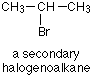
The SN2 mechanism is possible because the back of the molecule isn't completely cluttered by alkyl groups and so the approaching nucleophile can still get at the ![]() + carbon atom. The SN1 mechanism is possible because the secondary carbocation formed in the slow step is more stable than a primary one. It isn't as stable as a tertiary one though, and so the SN1 route isn't as effective as it is with tertiary halogenoalkanes.
+ carbon atom. The SN1 mechanism is possible because the secondary carbocation formed in the slow step is more stable than a primary one. It isn't as stable as a tertiary one though, and so the SN1 route isn't as effective as it is with tertiary halogenoalkanes.