
Hydroamination Reactions of Alkenes
- Page ID
- 62668

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Hydroamination reactions of alkenes represent additions of N-H bonds across carbon-carbon double bonds. Viable substrates for these reactions include unactivated alkenes, vinyl arenes, allenes, and strained alkenes. The scope of the nitrogen-containing reactant includes amines, azoles, and N-protected substrates. A variety of catalysts have been employed including alkali metals, alkaline earth metals, transition metals, and lanthanides.[1]
Introduction
As a consequence of the importance of nitrogen-containing compounds in biological and pharmaceutical applications, methods for the construction of carbon-nitrogen bonds have been heavily studied for at least 150 years. Hydroamination, the addition of HNR2 across a carbon-carbon π bond, is in principle one of the most atom-economical methods for the synthesis of this class of compounds. Catalytic hydroaminations of alkenes, allenes, and dienes (which may be inter- or intramolecular) afford amines, imines, and enamines (Eq. 1).
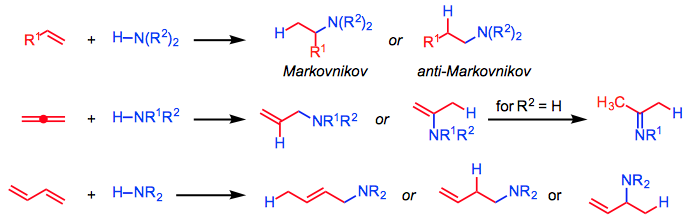
The scope of amine substrates includes ammonia, primary and secondary aliphatic and aromatic amines, azoles, hydrazines, and N-protected amines. Transition metal, rare earth, and alkali metal catalysts have been applied successfully, although the appropriate choice of catalyst depends on the unsaturated and amine substrates.
In general, hydroaminations present issues of site selectivity (Markovnikov/anti-Markovnikov) and enantioselectivity. Site selectivity is a function of the unsaturated substrate as well as the catalyst. The scope of enantioselective hydroamination is very broad; however, a particular catalyst system is typically limited to a fairly narrow range of substrates.
Mechanism and Stereochemistry
Prevailing Mechanisms
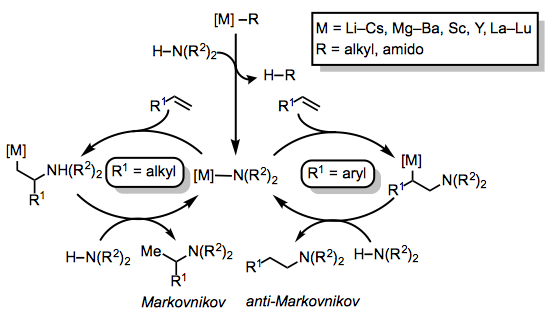
Mechanisms of alkene hydroaminations depend strongly on the catalyst system employed. Catalysts that include electropositive elements such as alkali, alkaline earth, and rare earth metals typically operate through a metal-amido species that undergoes nucleophilic addition to the alkene (Eq. 2).[2] The site selectivity of this addition depends on the substitution of the alkene: whereas aliphatic alkenes typically undergo Markovnikov addition, aromatic alkenes more commonly engage in anti-Markovnikov addition due to stabilization of the resulting benzylic metal intermediate.
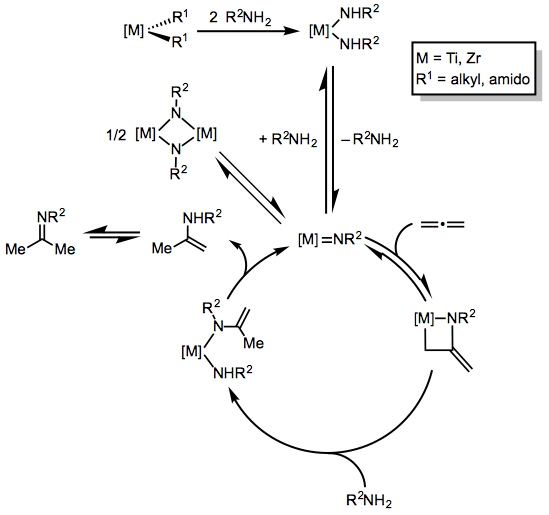
Hydroaminations of allenes catalyzed by Group 4 metals proceed by a different mechanism involving a metal imido intermediate (Eq. 3).[3] After formation of a bis-amido precursor, α-elimination generates the metal imido species, which engages in [2+2] cycloaddition with the allene substrate. Protonation then generates an enamido complex with an additional amido ligand; α-elimination then regenerates the metal-imido species.
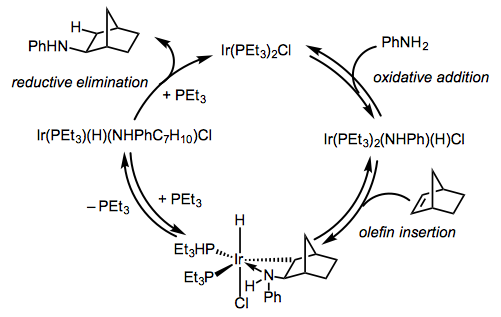
Mechanisms of hydroaminations catalyzed by late transition metals are not as well understood as the mechanisms discussed above. Nonetheless, it is generally believed that these reactions involve either amine activation via N-H insertion or alkene activation via π-coordination. Intermolecular hydroamination of norbornene catalyzed by an iridium(I) complex provides an example of the former (Eq. 4).[4] Oxidative addition of the amine is followed by insertion of the alkene and reductive elimination.
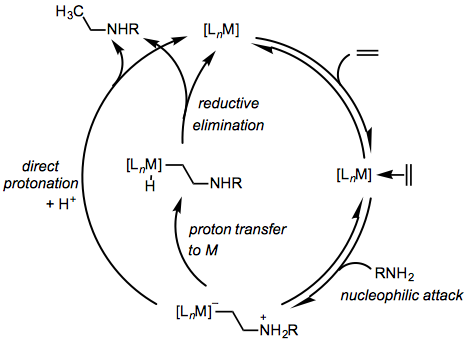
Group 9 and 10 metal complexes tend to react via an alkene activation pathway involving coordination of the alkene to the metal center and external attack of the amine on the coordinated alkene (Eq. 5).[5] Product formation may occur either by proton transfer to the metal center followed by reductive elimination or direct protonation of the β-aminoalkyl ligand.
Scope and Limitations
The scope of alkene hydroaminations is very broad and includes unactivated and activated alkenes as well as primary and secondary alkyl- and arylamines. However, this broad substrate scope is accompanied by an almost equally broad array of potential catalyst systems. Careful consideration must thus be given to the nature of the alkene and amine substrates in choosing the ideal catalyst. For example, late transition metal catalysts are generally incompatible with basic alkylamines and require either N-protected or aniline substrates.
Small unactivated alkenes generally require harsh conditions to undergo intermolecular hydroaminations, particularly when ammonia is used.[6] Catalyst decomposition is often a significant concern in reactions of alkylamines with unactivated alkenes. On the other hand, less basic anilines are more amenable to reaction with unactivated alkenes; for example, a catalyst system based on rhodium trichloride is effective in the reaction of ethylene with anilines (Eq. 6).[7]
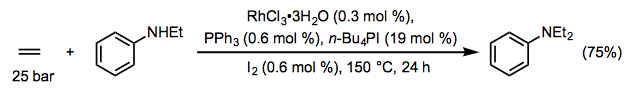 (6)
(6)
Unactivated alkenes are significantly more reactive in intramolecular hydroaminations and the scope of available catalysts is accordingly much broader. Rare earth metal catalysts are among the most active for intramolecular reactions of amino alkenes (Eq. 7).[8] Exclusive exo selectivity is observed in these reactions and in related reactions employing alkali or alkaline earth metal catalysts.
(7)

Early transition metal catalysts are also effective in intramolecular hydroaminations of amino alkenes (Eq. 8).[9] In this case, cyclization occurs efficiently even in the absence of geminal substituents in the tethering alkyl chain.
(8)
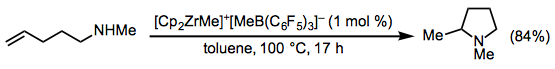
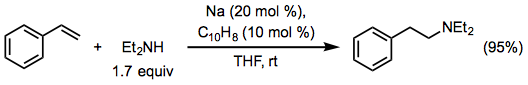
Conjugation significantly increases the reactivity of the alkene in vinyl arenes, although control of site selectivity can be difficult. For example, sodium metal catalyzes the addition of primary or secondary aliphatic amines to styrene with anti-Markovnikov selectivity (Eq. 9).[10]
(9)
Intramolecular reactions of vinyl arenes proceed similarly to reactions of unactivated amino alkenes described above. Catalysts based on rare earth metals are among the most active and react with exo selectivity (Eq. 10).[11]
(10)
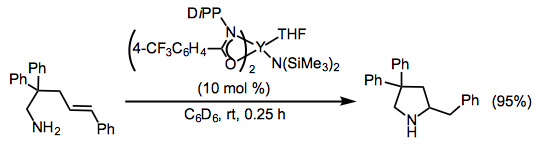
Although 1,3-dienes display comparable reactivity to vinyl arenes in hydroaminations, the possibility of 1,2- or 1,4-addition complicates this class of reactions. Late transition metal catalysts are known to catalyze the 1,4-addition of anilines to acyclic 1,3-dienes (Eq. 11).[12] Related reactions of alkylamines show a lack of site selectivity and issues with diene oligomerization.[13]
(11)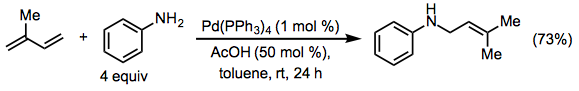
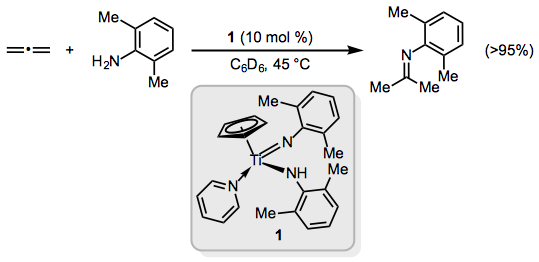
Allenes may undergo hydroamination to give enamines, imines, or allyl amines. Early transition metal catalysts such as the titanium half-sandwich imido complex 1 promote formation of imines exclusively (Eq. 12).[14]
(12)
Late transition metal catalysts are more active in the formation of allyl amines. For example, the gold(I) N-heterocyclic carbene complex 2 catalyzes the addition of N-protected amines to substituted allenes at room temperature (Eq. 13).[15]
(13)
Along with conjugated alkenes and cumulenes, strained alkenes represent a third class of substrates activated toward hydroamination. Like allenes, methylenecyclopropanes may afford imines, enamines, or allyl amines upon hydroamination. For instance, rare earth metal complexes catalyze the hydroamination of methylenecyclopropane to afford imines with ring opening (Eq. 14).[2]
(14)
In analogy to reactions of allenes, late transition metal catalysts react with substituted methylenecyclopropanes to afford allylamines (Eq. 15).[16]
(15)