
5.5: Reactions of Alkynes
- Page ID
- 354413

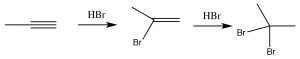
As you might predict, alkynes often behave in a similar way to alkenes. The triple-bonded carbons are an electron-rich region of the molecule and we would expect them to undergo electrophilic addition, in a similar manner to alkenes. So, for example, we see Markovikov addition across the triple bond with \(\mathrm{HBr}\) (\(\rightarrow\)), the only difference being that if excess \(\mathrm{HBr}\) is present, two—rather than one—bromine atom will be added; one to each of the originally triple-bonded carbons. Other reagents behave in a similar manner. For example \(\mathrm{Br}_{2}\) will also add across the triple bond to give first the dibromo, and then the tetrabromo compound.
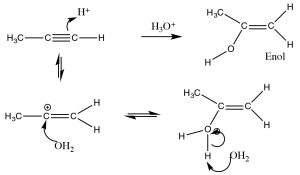
In contrast, when water is added across the triple bond we find a somewhat different outcome. While the initial steps are the same: the electrophile (\(\mathrm{H}^{+}\)) adds to the least-substituted carbon, and the nucleophile (\(\mathrm{H}_{2}\mathrm{O}\)) adds to the carbocation that is produced. This produces a new functionality called an enol (A combination of alkene and alcohol). The enol now undergoes what is known as a tautomerism: the proton from the alcohol moiety is removed (by water as a base), and another proton is picked up on the alkene \(\mathrm{CH}_{2}\) carbon (\(\rightarrow\)). As we have seen many times before this type protonation/deprotonation reaction occurs readily on either oxygen or nitrogen, but this is the first time we have seen it on a carbon; keto-enol tautomerism is an important part of the reactions of carbonyl groups.
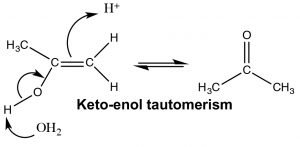
The keto and enol forms appear to be different compounds and we might be tempted to classify them as structural isomers—but they are not. The keto- and enol- forms always exist in an equilibrium with one another, and even though we usually write the structure with the carbonyl group (the keto form), there is always a small amount of the enol form present. The transition between keto- and enol- forms of the nucleotide bases initially confused Watson and Crick in their modeling of DNA structure.[7]
Reduction of alkynes:
Addition of hydrogen (\(\mathrm{H}_{2}\)) to alkynes can be accomplished in several ways. It is possible to completely reduce the alkyne to the corresponding fully-saturated alkane through the addition of two \(\mathrm{H}_{2}\) molecules. In fact, when using catalysts such as \(\mathrm{Pd}\) (palladium) or \(\mathrm{Pt}\) (platinum) the reaction cannot be stopped at the intermediate alkene stage. There are, however, specialized catalysts that allow for partial hydrogenation to the alkene. One of these is known as Lindlar’s catalyst, which is less efficient (poisoned) catalyst. As one might expect (by analogy with alkene reduction), the cis hydrogenated product is formed (\(\downarrow\)).
![]()
It is also possible to reduce an alkyne to the trans product, but to do this we have to use a different method; a method that involves the stepwise addition of the components of the reduction. The conditions for this reduction require a source of protons and a separate source of electrons. For example, a solution of sodium metal (a source of electrons) in liquid ammonia (a source of protons) at low temperature (since ammonia boils at \(-33^{\circ} \mathrm{C}\)), can be used to reduce an alkyne, but since the reaction proceeds via a stepwise (not concerted) addition, the product formed is the trans alkene: the most stable product (\(\rightarrow\)).
![]()
Acidity of Terminal Alkynes:
One alkyne-specific reaction involves the acidity of protons attached to sp hybridized carbons. The \(\mathrm{pK}_{\mathrm{a}\) of such protons is around \(25\), which is much lower than that of alkanes (\(> 55\)) or alkenes (\(\sim 45\)). In fact, “terminal” alkyne protons can be removed by strong bases such as \(\mathrm{NH}_{2}-\) (the amide ion), since the \(\mathrm{pK}_{\mathrm{a}\) of \(\mathrm{NH}_{3}\) (ammonia) is \(33\) (\(\downarrow\)).
![]()
The acidity of terminal alkyne protons can be explained by the idea that the negative charge (the lone pair on the resulting anion) is located in an sp hybrid orbital. The more “s” character in the hybrid orbital, the closer to the nucleus. Since the sp orbital is more “s” than either \(\mathrm{sp}^{2}\) or \(\mathrm{sp}^{3}\) orbitals, then the electron of the carbon anion is held closer to the nucleus and is therefore more stable than if the carbon were \(\mathrm{sp}^{2}\) or \(\mathrm{sp}^{3}\) hybridized. The effect is similar to the effective nuclear charge explanation for the trends in electronegativity across the periodic table (i.e. why fluorine is more electronegative than oxygen). The alkyne anion is very useful because it is now a carbon nucleophile, and will attack electrophilic carbon species in an \(\mathrm{S}_{\mathrm{N} 2\) reaction. This is the first example we have seen of carbon carbon bond formation (although we will see many more).
![]()