5.1: Reactions of Alkenes- Electrophilic Addition
- Page ID
- 354360

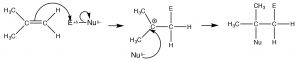
The double-bonded carbons of an alkene are electron-rich, that is, the electron density is high in the region of the double bond. Therefore, the “signature” reaction of alkenes involves initial attack on an electrophile. Instead of a substitution, alkenes undergo electrophilic addition, a reaction in which a two-component reactant adds across the double bond. The reaction begins with an electrophilic attack by the double bond onto the reactant which produces a carbocation that then undergoes nucleophilic attack. In the case of unsymmetrical alkenes (where the groups attached to the double-bonded carbons are not exactly the same), the most stable carbocation is produced. This reaction is regioselective, that is, we can predict the orientation of reactant addition across the double bond. If we designate the reagent as \(\mathrm{E}\) (for electrophile) or \(\mathrm{N}\) (for nucleophile), the reaction would proceed as outlined below.
| Reagent | Electrophile | Nucleophile | Typical Conditions |
| \(\mathrm{HBr}\) | \(\mathrm{H}^{+}\) | \(\mathrm{Br}^{-}\) | Low Temp |
| \(\mathrm{H}_{2}\mathrm{O}\) | \(\mathrm{H}^{+}\) | \(\mathrm{H}_{2}\mathrm{O}\) (with loss of \(\mathrm{H}^{+}\) after addition) | Aqueous acid/Low Temp |
| \(\mathrm{ROH}\) | \(\mathrm{H}^{+}\) | \(\mathrm{ROH}\) | \(\mathrm{ROH} / \mathrm{H}_{3} \mathrm{O}^{+}\) |
| \(\mathrm{Br}_{2}\) | \(\mathrm{Br}^{+}\) | \(\mathrm{Br}^{-}\) | \(\mathrm{Br}_{2} / \mathrm{CCl}_{4}\) |
| \(\mathrm{BrOH}\) | \(\mathrm{Br}^{-}\) | \({}^{-} \mathrm{OH}\) | \(\mathrm{Br}_{2} / \mathrm{H}_{2} \mathrm{O}\) |
| \(\mathrm{BrOR}\) | \(\mathrm{Br}^{+}\) | \({}^{-} \mathrm{OR}\) | \(\mathrm{Br}_{2} / \mathrm{ROH}\) |
The intermediate carbocation is the tertiary carbocation, (rather than the primary carbocation that would be produced by addition to the \(\mathrm{=CH}_{2}\) end of the double bond). This pattern of reaction is referred to as Markovinkov addition, after the person[1] who first discovered that \(\mathrm{HBr}\) adds in this way to a double bond. We can classify many reagents as combinations of electrophile and nucleophile and, in this way, predict how they will add across the double bond. Examples of such reagents are shown (\(\uparrow\)). Rather than memorizing the product of every type of addition across a double bond, it is much more productive to write a mechanism by determining which part is the electrophile, adding it to give the most stable carbocation, followed by the nucleophile.
Additions to alkenes are reversible:
Let us now take a closer look at the addition of water across a double bond. Such a reaction can be accomplished by reacting the alkene with dilute sulfuric acid at low temperatures. The first step is addition of a proton to produce the most stable carbocation—which is then attacked by water (the nucleophile). The final product is the alcohol that forms after a proton is transferred to water. In this case, we can consider the proton (or more accurately \(\mathrm{H}_{3}\mathrm{O}^{+}\)) as a catalyst since it is regenerated at the end of the reaction sequence.
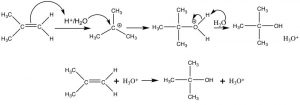
Acid-catalyzed addition of water across a double bond
At this point you might be asking yourself: well didn’t we just talk about the reverse reaction—that is, the elimination of \(\mathrm{H}_{2}\mathrm{O}\) from alcohols to give alkenes? Indeed we did! Many organic reactions are reversible[2], it is just a matter of manipulating the conditions. The exact reaction conditions will determine which reaction is favored.
As is the case with most addition reactions, the addition of water across an alkene is exothermic, that is, \(\Delta \mathrm{H}\) is negative because stronger (sigma) bonds are formed during the reaction and energy is released into the environment. A typical energy diagram is shown below.
Reaction energy diagram for addition/elimination across a double bond.
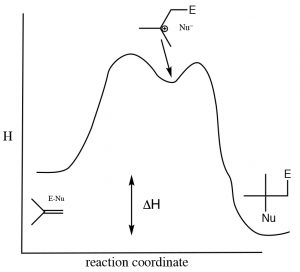
This means that \(\Delta \mathrm{H}\) for the elimination reaction must be positive (i.e. going from right to left on the diagram above). The question then is: why does an elimination reaction ever occur? To answer that, we have to recall that the thermodynamic criterion for a reaction to proceed is not simply a negative enthalpy change, but rather a negative change in the Gibbs change (\(\Delta \mathrm{G}\)). Recall that \(\Delta \mathrm{G}=\Delta \mathrm{H}-\mathrm{T} \Delta \mathrm{S}\). The change in entropy also influences the thermodynamic favorability of a reaction. In the elimination reaction, two molecules (alkene and water) are produced one alcohol molecule –the entropy change will be (Recall that entropy is associated with the number of possible arrangements of the system. Since two molecules will have more possible arrangements than one, this reaction will always be accompanied by an increase in entropy of the system). To encourage the equilibrium to shift to the right (the addition reaction) we need increase the temperature, which will increase the magnitude of the \(-\mathrm{T} \Delta \mathrm{S}\)\) term, making \(\Delta \mathrm{G}\) more negative (assuming that \(\Delta \mathrm{S}\) is positive). In general, additions to double bonds are carried out at lower temperatures, while elimination reactions involve heating the reaction solution. Another way to influence the equilibrium state is to change the relative concentrations of reactants or products. Because water is a reactant, increasing the concentration of water shifts the equilibrium position towards the addition product while lowering the water concentration favors the elimination reaction.

Specific reagents for additions across a double bond that reduce the carbocation problem
The problem with many of these simple addition reactions to a double bond is that they generate carbocations, which as we have seen already can lead to further reactions, resulting in skeletal rearrangements and the production of racemic mixtures (rather than a single stereoisomer). To address this issue, a number of reagents have been developed that minimize this problem. For example, a reagent that involves mercuric acetate (\(\mathrm{Hg}(\mathrm{OAc})_{2}\)) and sodium borohydride (\(\mathrm{NaBH}_{4}\)) as an intermediate can be used to add \(\mathrm{H}_{2}\mathrm{O}\), (or alcohol) across a double bond (\(\downarrow\)).
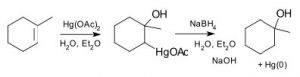
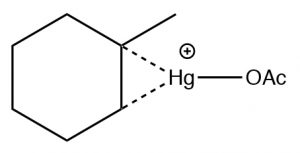
The reaction involves a mercury-stabilized cation (\(\rightarrow\)) that prevents unwanted rearrangements. The product is still a Markovnikov product (see above) but is often formed more cleanly, that is, without unwanted alternatives.
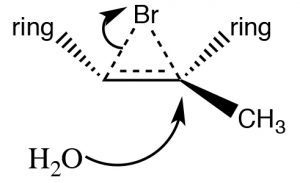
Another set of reactions that can be used to constrain molecular rearrangements and lead to stereospecific products are those that begin with the addition of bromine across the double bond. The simplest of these co-reactions is addition of \(\mathrm{Br}_{2}\) itself; since \(\mathrm{Br}\) is a large polarizable atom, the bromine molecule can become polarized and interact with the double bond as shown (\(\downarrow\)) to form a bromonium ion (rather than a carbocation).
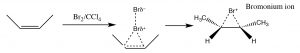
The bromonium ion can now undergo nucleophilic attack at either carbon (since in this example they are the same, that is, they are attached to identical groups), to produce the trans-dibromo addition product. The trans product is formed because the second step is an \(\mathrm{S}_{\mathrm{N}} 2\) reaction with the bromide nucleophile attacking the carbon from the back-side.
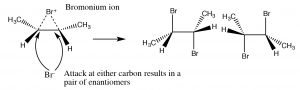
Addition of \(\mathrm{Br}_{2}\) is accomplished by using a reaction solvent such as carbon tetrachloride that does not interfere with the reaction. If water or an alcohol is used as the solvent, then attack on the bromonium ion comes from the solvent acting as the nucleophile in the second step.
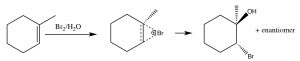
Again, the addition is trans, but now an incoming nucleophile (\(\mathrm{H}_{2}\mathrm{O}\)) will attack the carbon that is most carbocation-like, that is it is the most stabilized, as shown here \(\rightarrow\). The reaction is both regiospecific and stereospecific.