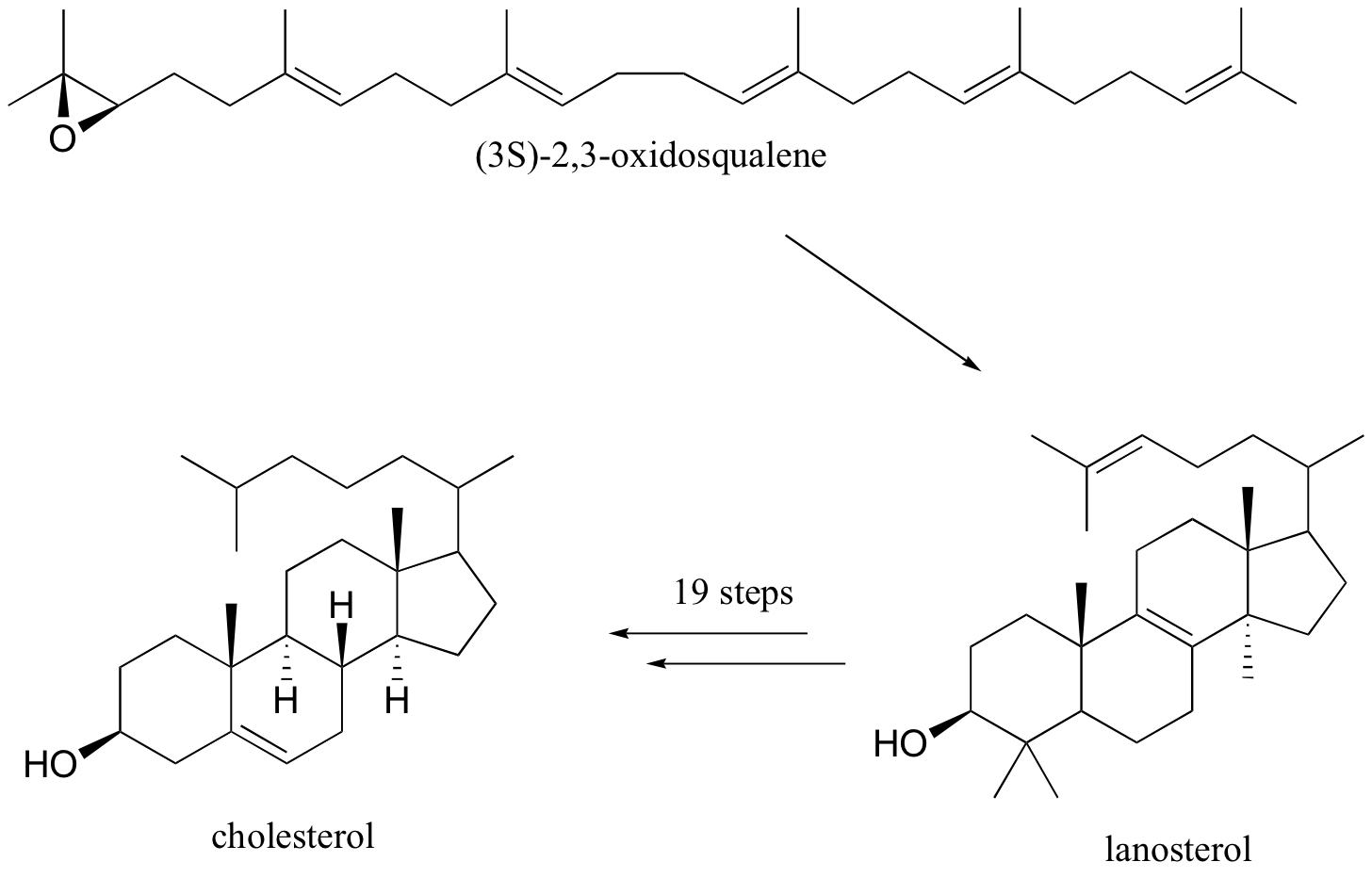
The terpenoids (aka isoprenoids) are a large (estimated 60% of known natural products ) and diverse group of lipids derived from five-carbon isoprene units assembled in thousands of combinations. Technically a terpenoid contains oxygen, while a terpene is a hydrocarbon. Often the two terms are used to refer collectively to both groups.
Isoprene Rule
Compounds classified as terpenes constitute what is arguably the largest and most diverse class of natural products. A majority of these compounds are found only in plants, but some of the larger and more complex terpenes (e.g. squalene & lanosterol) occur in animals. Terpenes incorporating most of the common functional groups are known, so this does not provide a useful means of classification. Instead, the number and structural organization of carbons is a definitive characteristic. Terpenes may be considered to be made up of isoprene (more accurately isopentane) units, an empirical feature known as the isoprene rule. Because of this, terpenes usually have \(5n\) carbon atoms (\(n\) is an integer), and are subdivided as follows:
| Classification |
Isoprene Units |
Carbon Atoms |
| monoterpenes |
2 |
C10 |
| sesquiterpenes |
3 |
C15 |
| diterpenes |
4 |
C20 |
| sesterterpenes |
5 |
C25 |
| triterpenes |
6 |
C30 |
Isoprene itself, a C5H8 gaseous hydrocarbon, is emitted by the leaves of various plants as a natural byproduct of plant metabolism. Next to methane it is the most common volatile organic compound found in the atmosphere. Examples of C10 and higher terpenes, representing the four most common classes are shown in the following diagrams. Most terpenes may be structurally dissected into isopentane segments. How this is done can be seen in the diagram directly below.
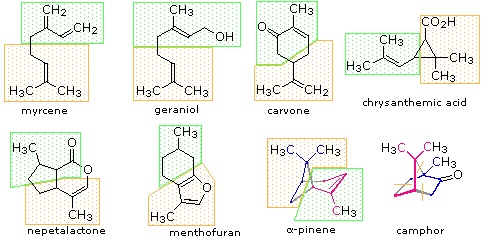
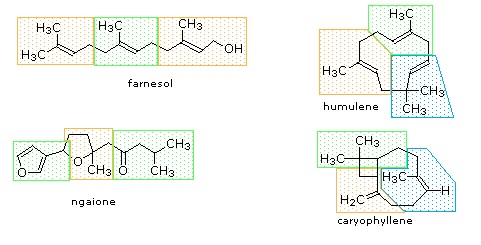 Figure 27.5.1: Monoterpenes and sesquiterpenes
Figure 27.5.1: Monoterpenes and sesquiterpenes
The isopentane units in most of these terpenes are easy to discern, and are defined by the shaded areas. In the case of the monoterpene camphor, the units overlap to such a degree it is easier to distinguish them by coloring the carbon chains. This is also done for alpha-pinene. In the case of the triterpene lanosterol we see an interesting deviation from the isoprene rule. This thirty carbon compound is clearly a terpene, and four of the six isopentane units can be identified. However, the ten carbons in center of the molecule cannot be dissected in this manner. Evidence exists that the two methyl groups circled in magenta and light blue have moved from their original isoprenoid locations (marked by small circles of the same color) to their present location. This rearrangement is described in the biosynthesis section. Similar alkyl group rearrangements account for other terpenes that do not strictly follow the isoprene rule.
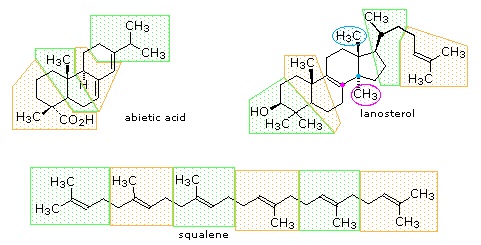 Figure 27.5.2: Triterpenes
Figure 27.5.2: Triterpenes
Polymeric isoprenoid hydrocarbons have also been identified. Rubber is undoubtedly the best known and most widely used compound of this kind. It occurs as a colloidal suspension called latex in a number of plants, ranging from the dandelion to the rubber tree (Hevea brasiliensis). Rubber is a polyene, and exhibits all the expected reactions of the C=C function. Bromine, hydrogen chloride and hydrogen all add with a stoichiometry of one molar equivalent per isoprene unit. Ozonolysis of rubber generates a mixture of levulinic acid ( \(CH_3COCH_2CH_2CO_2H\) ) and the corresponding aldehyde. Pyrolysis of rubber produces the diene isoprene along with other products.
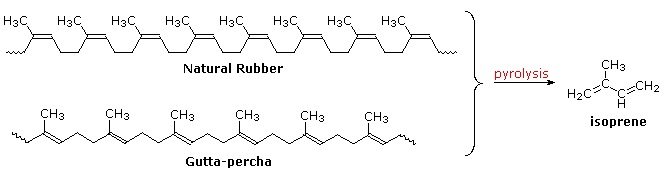
The double bonds in rubber all have a Z-configuration, which causes this macromolecule to adopt a kinked or coiled conformation. This is reflected in the physical properties of rubber. Despite its high molecular weight (about one million), crude latex rubber is a soft, sticky, elastic substance. Chemical modification of this material is normal for commercial applications. Gutta-percha (structure above) is a naturally occurring E-isomer of rubber. Here the hydrocarbon chains adopt a uniform zig-zag or rod like conformation, which produces a more rigid and tough substance. Uses of gutta-percha include electrical insulation and the covering of golf balls.
Mevalonate Pathway
Step 1 - Claisen Condensation
An early step in the biosynthesis of cholesterol and other ‘isoprenoid’ compounds is a Claisen condensation between two acetyl CoA molecules. An initial trans-thioesterase process transfers the acetyl group of the first acetyl CoA to an enzymatic cysteine (Reaction 1). In the Claisen condensation phase of the reaction, the alpha-carbon of a second acetyl CoA is deprotonated, forming an enolate (Reaction 2).

The enolate carbon attacks the electrophilic thioester carbon, forming a tetrahedral intermediate (Reaction 3) which quickly collapses to expel the cysteine thiol (Reaction 4) and produce acetoacetyl CoA.
Step 2 - Aldol Condensation
Acetyl CoA then reacts with the acetoacetyl CoA in an aldol-like addition. Subsequent hydrolysis produces (3S)-3-hydroxy-3-methylglutaryl CoA (HMG-CoA).
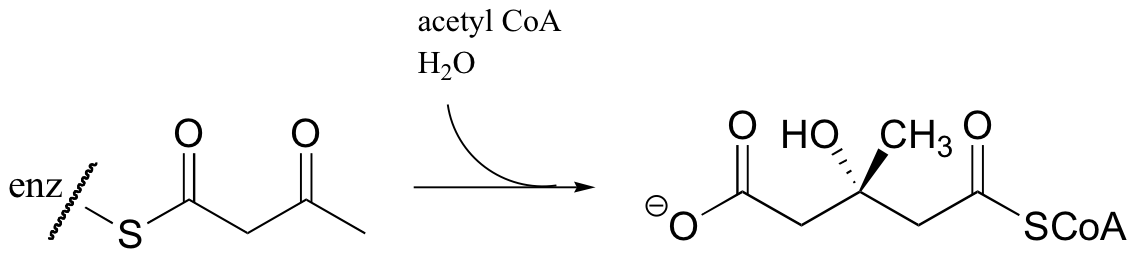
Generating HMG-CoA
Step 3 - Reduction of the Thioester
The thioester is reduced first to an aldehyde, then to a primary alcohol by two equivalents of NADPH producing (R)-mevalonate. The enzyme catalyzing this reaction is the target of the statin family of cholesterol-lowering drugs.
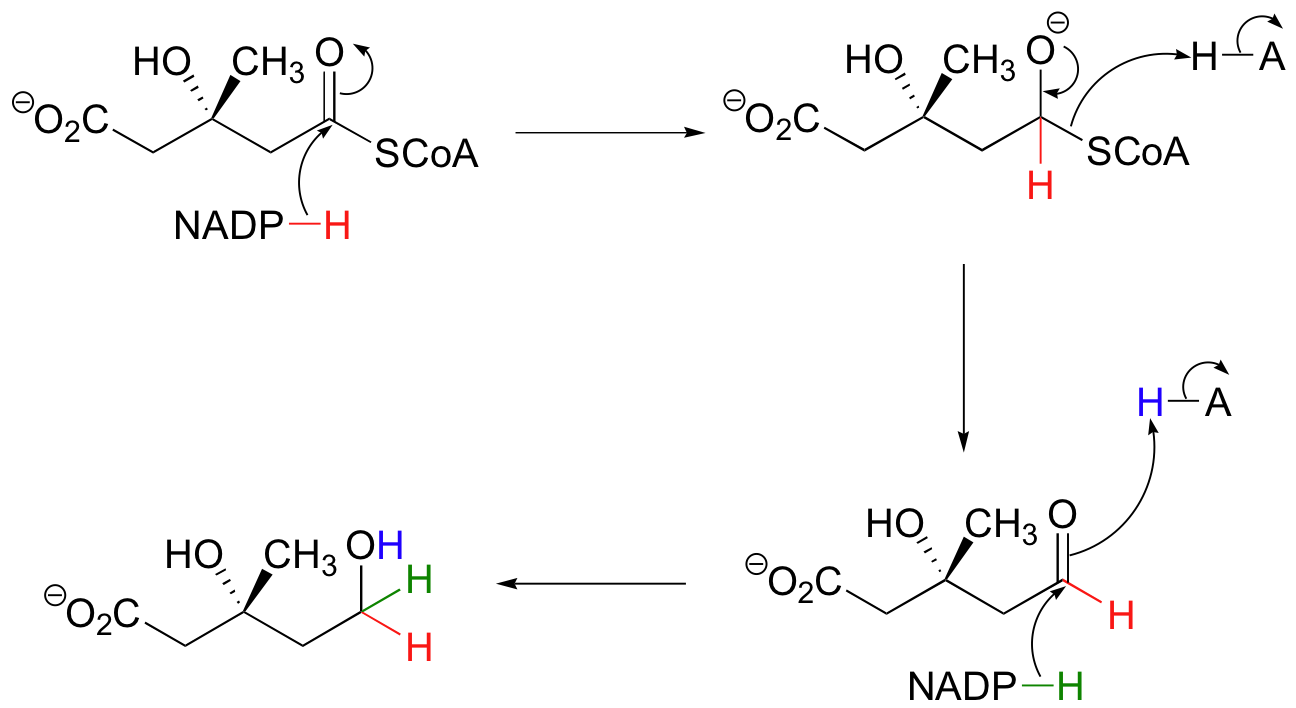
Generating (R)-Mevalonate
Step 4 - Mevalonate Phosphorylation
Two phsophorylations by adenosine triphosphate (ATP) occur at the terminal hydroxyl/phosphorus group through nucleophilic substitution, followed by a third ATP phosphorylation of the tertiary hydroxyl group.
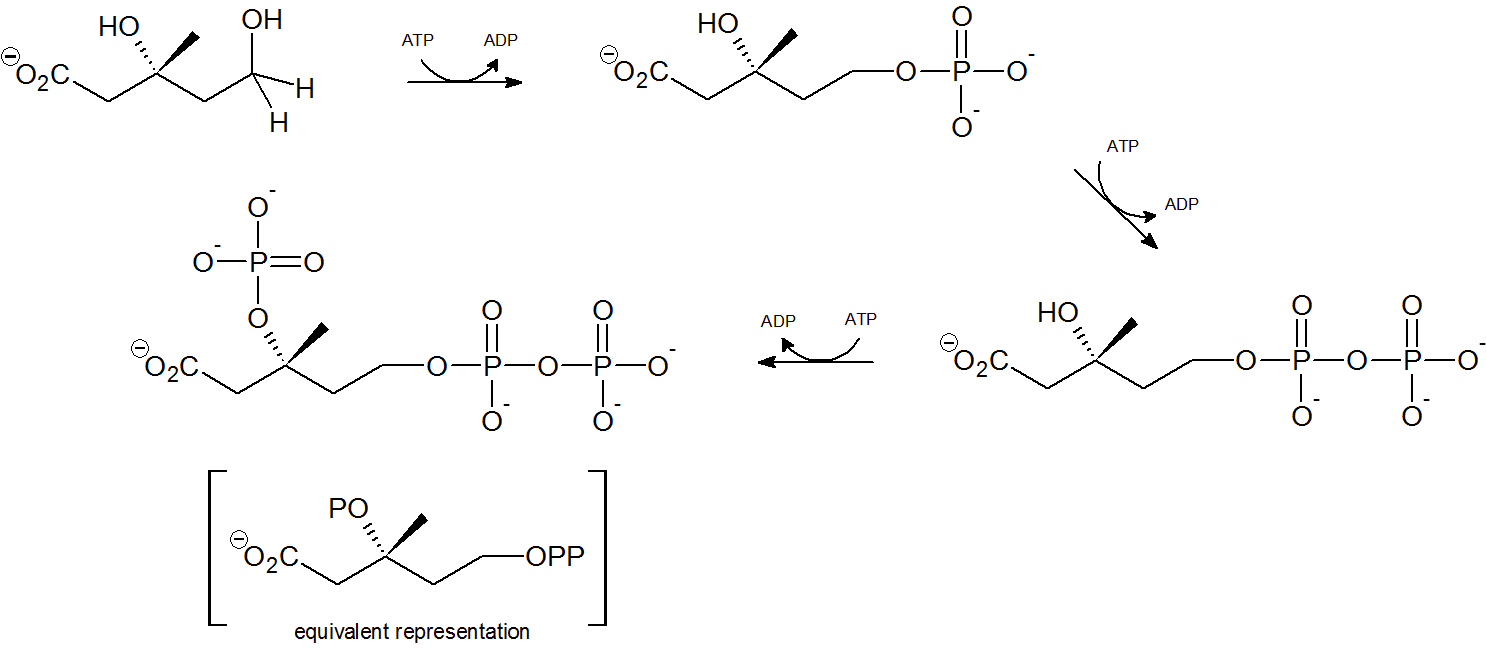
Step 5 - Decarboxylation
Finally isopentenyl diphosphate (IPP), the 'building block' for all isoprenoid compounds, is formed from a decarboxylation-elimination reaction.
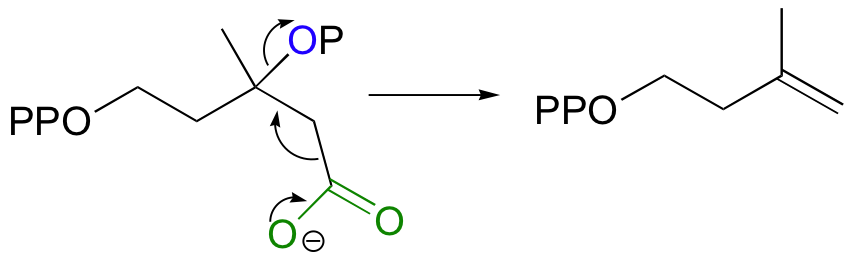
Conversion of IPP to Terpenoids
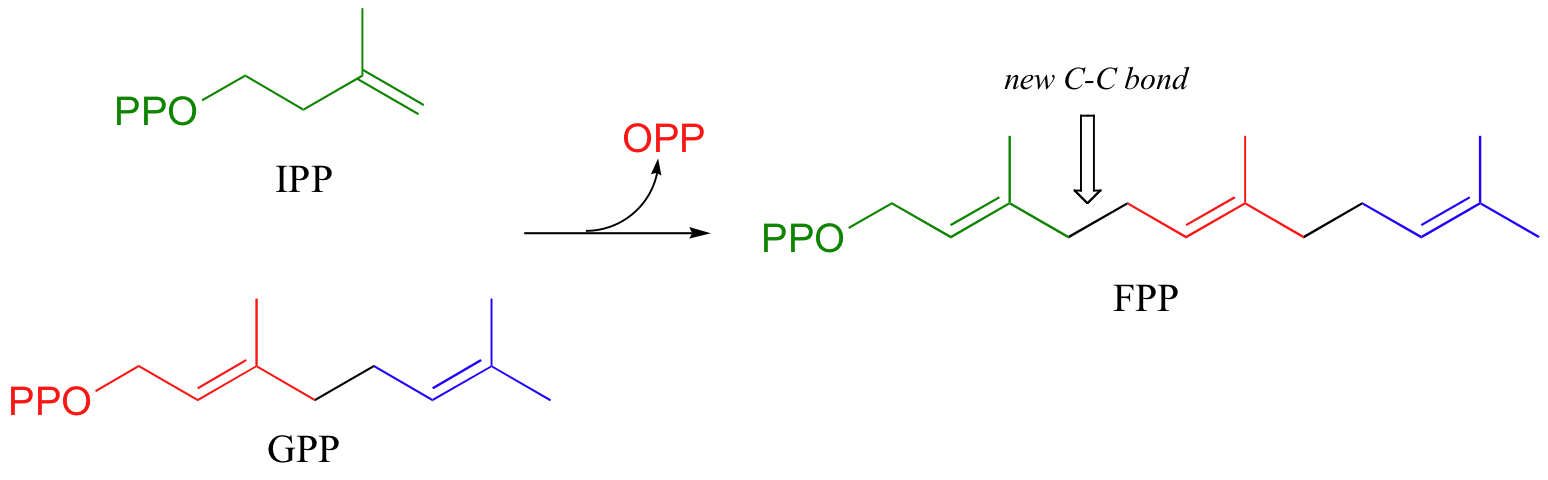
The electrophilic double bond isomerization catalyzed by IPP isomerase is a highly reversible reaction, with an equilibrium IPP:DMAPP ratio of about 6:1. In the next step of isoprenoid biosynthesis, the two five-carbon isomers condense to form a 10-carbon isoprenoid product called geranyl diphosphate (GPP).
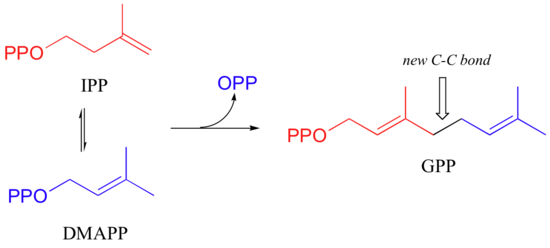
This is a nice example of an electrophilic addition/elimination mechanism:
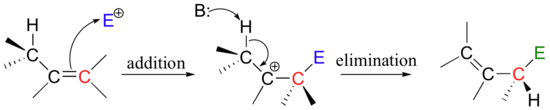
The first step is ionization of the electrophile - in other words, the leaving group departs and a carbocation intermediate is formed. In this case, the pyrophosphate group on DMAPP is the leaving group, and the electrophilic species is the resulting allylic carbocation.
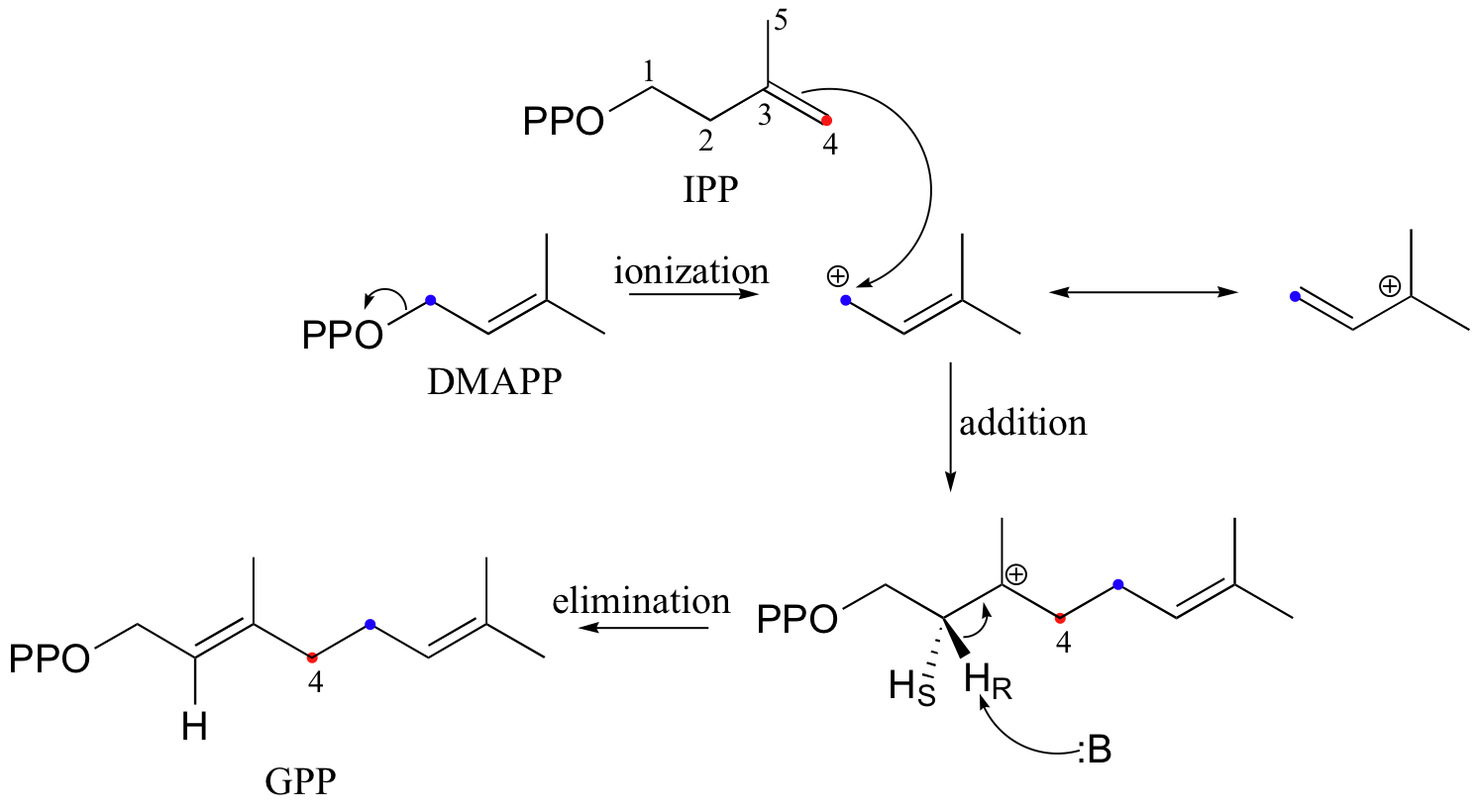
In the condensation (addition) step, the C3-C4 double bond in IPP attacks the positively-charged C1 of DMAPP, resulting in a new carbon-carbon bond and a second carbocation intermediate, this time at a tertiary carbon. In the elimination phase, proton abstraction leads to re-establishment of a double bond in the GPP product. Notice that the enzyme specifically takes the pro-R proton in this step.
To continue the chain elongation process, another IPP molecule can then condense, in a very similar reaction, with C1 of geranyl diphosphate to form a 15-carbon product called farnesyl diphosphate (FPP).
How do we know that these are indeed SN1-like mechanisms with carbocation intermediates, rather than concerted SN2-like mechanisms? First of all, recall that the question of whether a substitution is dissociative (SN1-like) or associative (SN2-like) is not always clear-cut - it could be somewhere in between, like the protein prenyltransferase reaction. The protein prenyltransferase reaction and the isoprenoid chain elongation reactions are very similar: the electrophile is the same, but in the former the nucleophile is a thiolate, while in the latter the nucleophile is a pi bond.
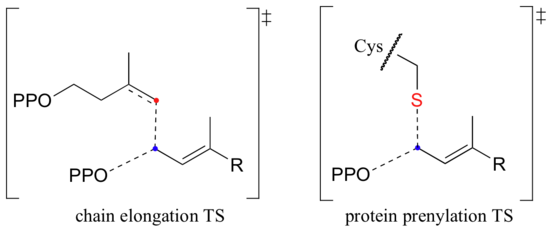
This difference in the identity of the nucleophilic species would lead one to predict that the chain elongation reaction has more SN1-like character than the protein prenylation reaction. A thiolate is a very powerful nucleophile, and thus is able to push the pyrophosphate leaving group off, implying some degree of SN2 character. The electrons in a pi bond, in contrast, are only weakly nucleophilic, and thus need to be pulled in by a powerful electrophile - ie. a carbocation.
So it makes perfect sense that the chain elongation reaction should more SN1-like than SN2-like. Is this in fact the case? We know how to answer this question experimentally - just run the reaction with fluorinated DMAPP or GPP substrates and observe how much the fluorines slow things down.
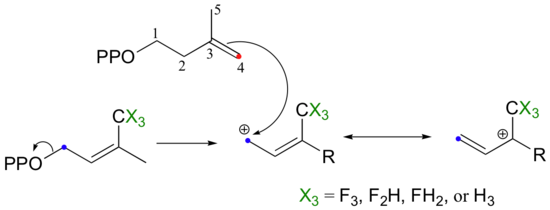
If the reaction is SN1-like, the electron-withdrawing fluorines should destabilize the allylic carbocation intermediate and thus slow the reaction down considerably. If the mechanism is SN2-like, the fluorine substitutions should not have a noticeable effect, because a carbocation intermediate would not be formed. When this experiment was performed with FPP synthase, the results were dramatic: the presence of a single fluorine slowed down the rate of the reaction by a factor of about 60, while two and three fluorines resulted in a reaction that was 500,000 and 3 million times slower, respectively (J. Am. Chem. Soc. 1981, 103, 3926.) These results strongly suggest indicate the formation of a carbocation intermediate in an SN1-like displacement.
In this section, we will briefly examine the reaction catalyzed by an enzyme called squalene synthase, an important enzymatic transformation that involves some very interesting and unusual electrophilic additions, rearrangements, and reactive intermediates. This particular enzyme is also of interest because it represents a potential new target for cholesterol-lowering drugs.
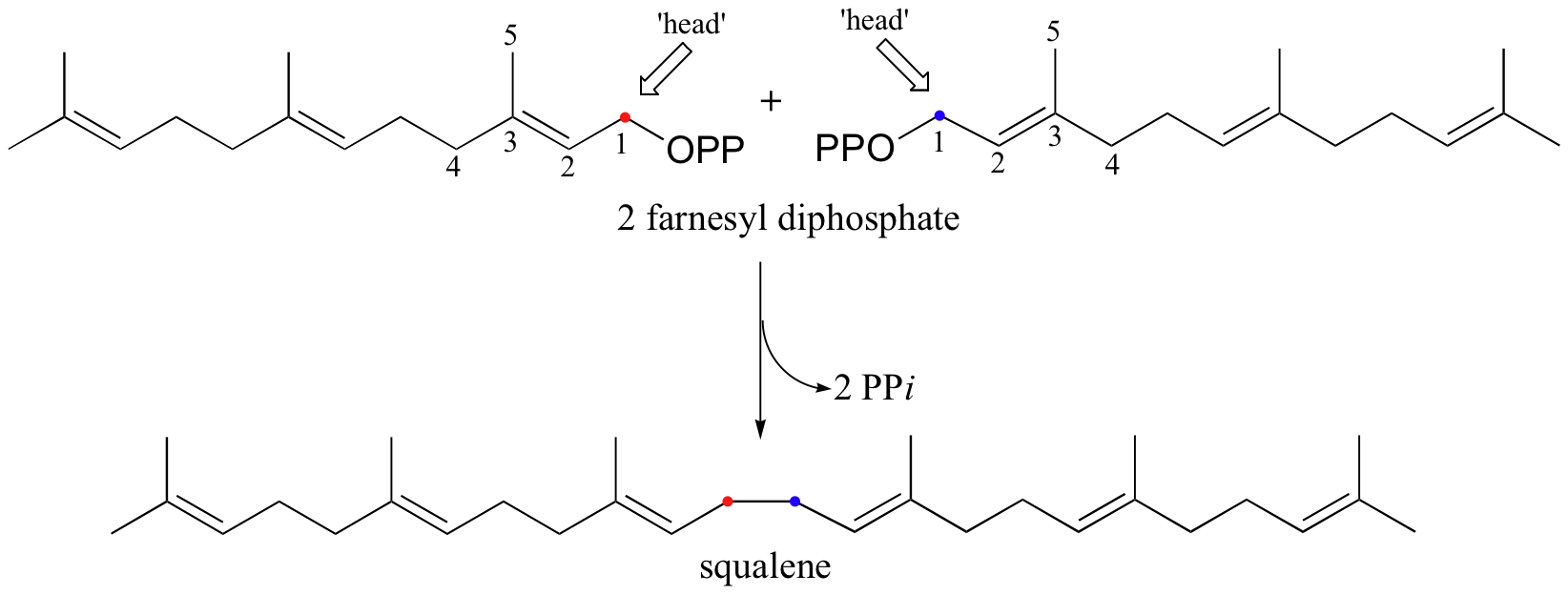
Cholesterol, as we discussed earlier in this chapter, is derived from a 30-carbon isoprenoid molecule called squalene. Squalene, in turn, is derived from the condensation of two molecules of farnesyl diphosphate (FPP), a 15-carbon isoprenoid. You may recall that FPP is the product of the C4 to C1, or 'head to tail' electrophilic condensation of isoprenoid chains:
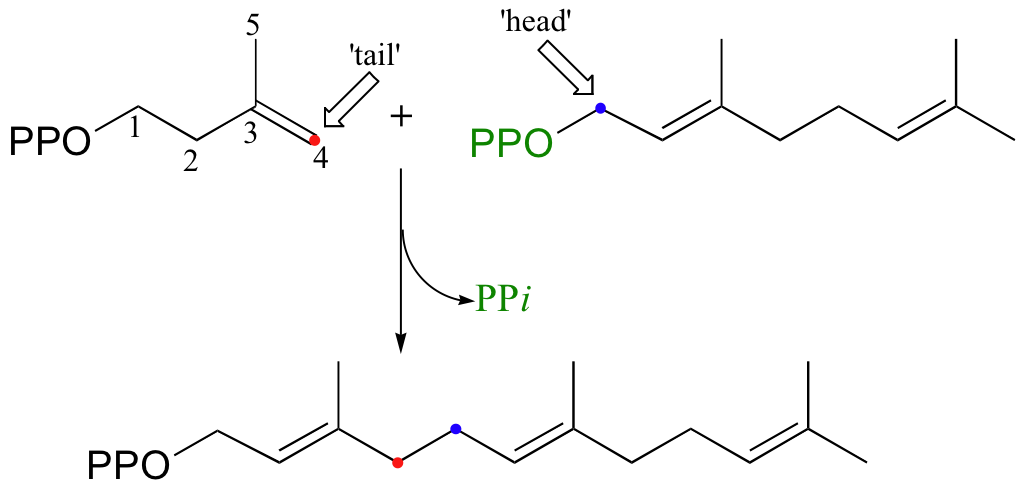
The condensation of two molecules of FPP to form squalene, however, is something different: this is a 'head to head' condensation, where C1 of the first molecule forms a bond to C1 of the second. The chemistry involved is quite a bit more complicated.
The first two steps are familiar: first, the pyrophosphate on one FPP molecule leaves (step 1), resulting in an allylic carbocation that is attacked by the C2-C3 π bond of the second molecule (step 2).
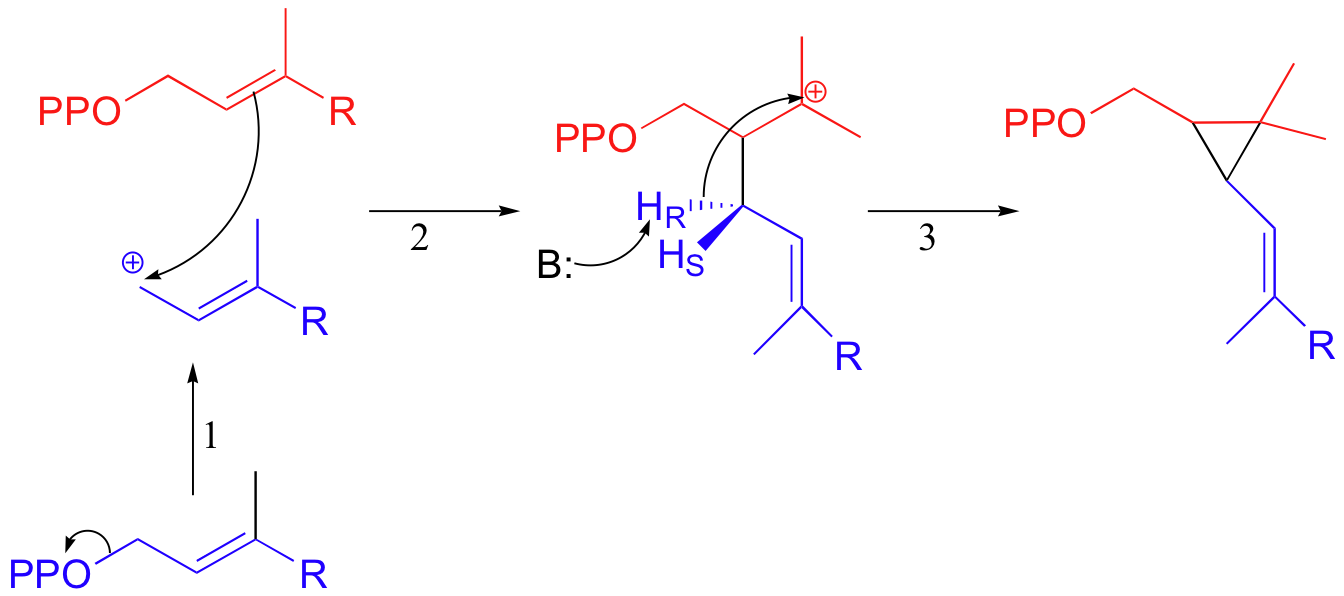
This results in a new carbon-carbon bond between the two FPP molecules, but with incorrect C1 to C2 connectivity (remember, the overall reaction is a C1 to C1 condensation). In step 3, a proton is abstracted and the electrons from the broken C-H bond bridge across a 2-carbon gap to form a cyclopropyl intermediate.
In the second stage of squalene synthesis, the second pyrophosphate group leaves, generating a cyclopropylcarbinyl cation (step 4). Because this is a primary carbocation, you probably are wondering about how stable it could be (and thus how likely an intermediate). As it turns out, such carbocations are remarkably stable, due to favorable interactions between the empty orbital and orbitals on the three-membered ring (the level of bonding theory needed to really understand this idea is beyond the scope of this text, but you may learn about it if you take a class in advanced organic chemistry). What occurs next is an alkyl shift leading to a tertiary carbocation (step 5).
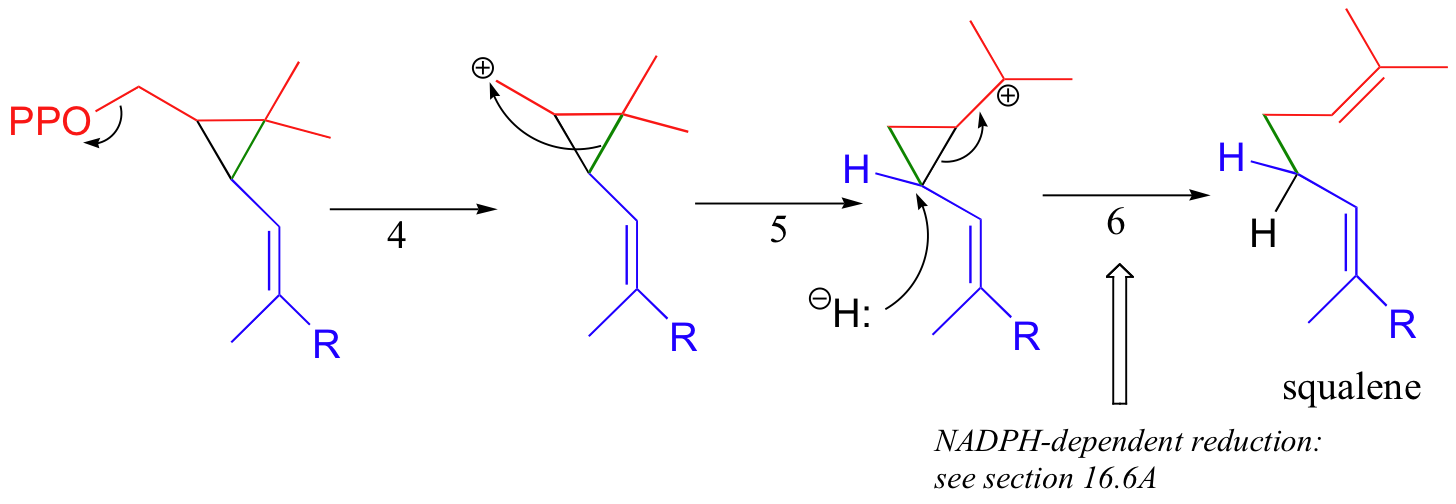
Discussion of the final step (step 6) will need to be put off - this is a reduction with a hydride nucleophile derived from a coenzyme called NADPH. Although this may seem like an extremely convoluted (and perhaps unlikely!) mechanism, there is much experimental evidence to back it up.