6.5: Quinine
- Page ID
- 285464

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis of Alkaloids from Secologanin
A comparison of the structures of polycyclic alkaloids with a variety of topologically different skeletons, such as preakuammicine (317), catharanthine (318), and tabersonine (319), suggests a biosynthetic strategy which assembles these heteromulticycles by the union of an aminoethyl indole starting material 400 with ten additional skeletal carbons. The aminoethyl indole, tryptamine (400), is reasonably derived from decarboxylation of the amino acid L-tryptophan (259) by a process analogous to the decarboxylation of tyrosine (7) discussed in section 6.4.
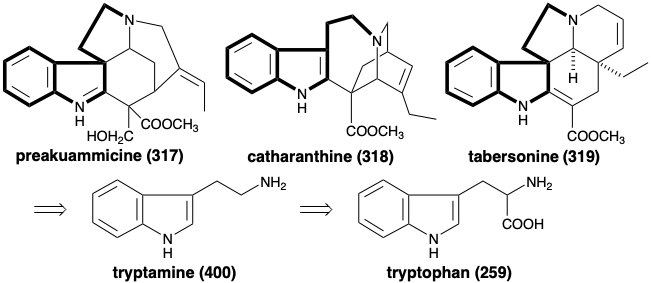
The origin of the remaining ten skeletal carbons is less obvious. The remarkable fact that these remaining carbons have a common origin is nicely illustrated by studies on the time evolution of alkaloid production in germinating seedlings of Vinca Rosea.16 Thus, the alkaloids 317 - 326, and 328 are all isolable from this plant while 327 is a putative common intermediate for the generation of 318 and 319 by two different 2\(\pi\) + 4\(\pi\) cycloadditions.
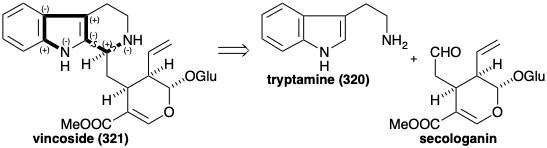
In the early intermediate vincoside (321), there is a consonant circuit that connects the two nitrogens. Two polar disconnections in this circuit reveal an aldehyde precursor that bears little resemblence to a monoterpene besides its ten skeletal carbon atoms. Nevertheless, this aldehyde is secologanin whose terpenoid origin was discussed in chapter 4 (see section 4.4).
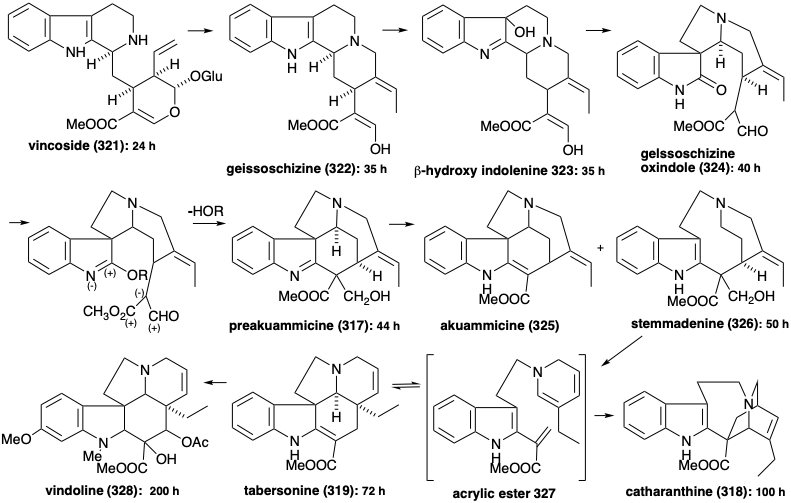
The biosynthesis of over 1,000 indole alkaloids from tryptophan (259) begins with a Mannich reaction between tryptamine (320) and secologanin to produce vincoside (321). Hydrolysis of the glucoside 321 and deketalization of the resulting hemiketal 329 affords amino-aldehyde 330. Cyclization of the latter gives an iminium derivative 331. Intramolecular Michael addition of an oxygen nucleophile followed by reduction affords another isolable product, ajmalicine (332). Alternatively, reduction of 331 affords 322.
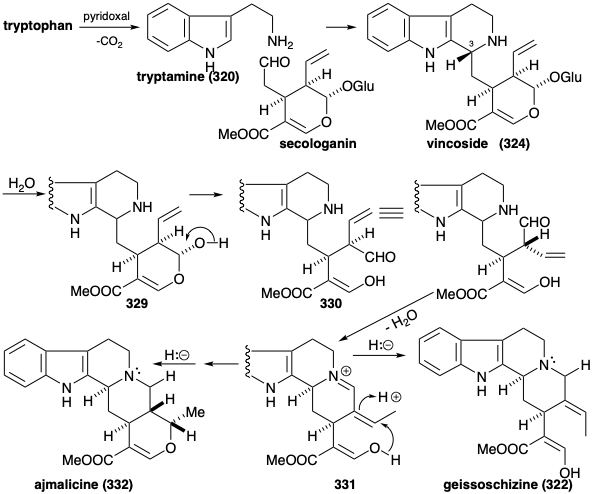
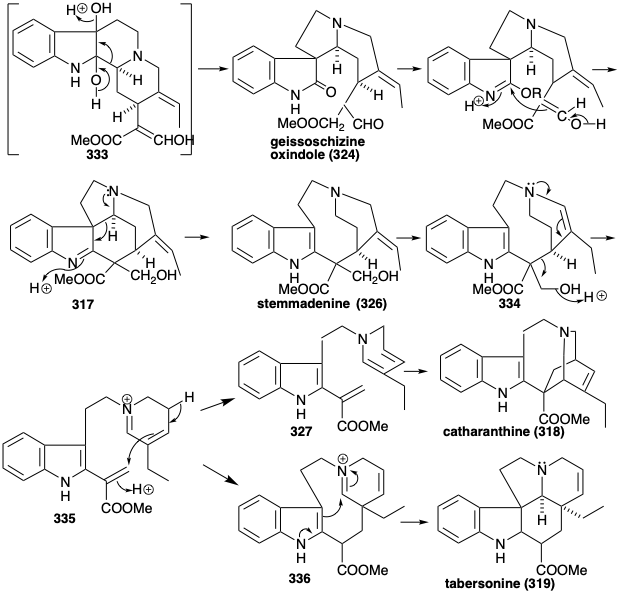
Generation of the other skeletal types from 322 involves rearrangements that are enabled by oxidative introduction of a vicinal diol array to produce 333. This vicinal hydroxylation is accomplished by a stepwise process through the hydration of an isolable intermediate, the β-hydroxyindolenine 323. A pinacol rearrangement of 333 produces 324. Conversion to an imino ester would endow 324 with the reactivity required to form preakuammicine (317) by cyclization and reduction. Retero-aldol-like fragmentation of 317 followed by the reduction of the resulting immine affords stemmadenine (326). A second fragmentation of the enamine isomer 334 of 326 apparently produces an acrylic ester intermediate 335. The dienamine tautomer 327 of 335 provides the iboga alkaloid skeleton of catharanthine (318) by an intramolecular Diels-Alder reaction (not necessarily concerted). Alternatively, the aspidosperma alkaloid skeleton of tabersonine (319) arises from the acrylic ester 335 via polyene cyclization to 336 and subsequent aldol-like cyclization of the latter followed by proton loss to afford tabersonine (319).
Biosynthesis of Quinine
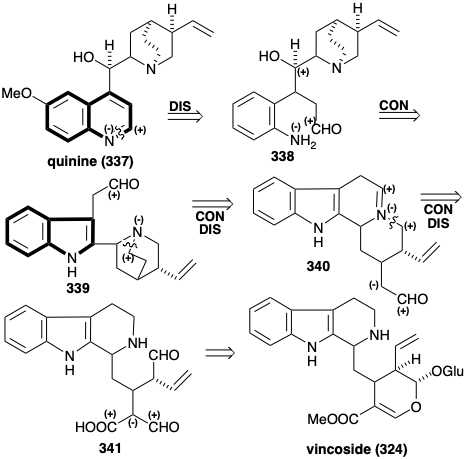
Even more cryptic is the biosynthesis of several alkaloids containing the quinoline heterocyclic ring system such as quinine (337). The surprising fact that the carbon skeletons of these alkaloids are also derived from tryptophan and secologanin further illustrates the lengths to which Nature must go to achieve the biosynthesis of some natural products owing to a limited inventory of available starting materials. It is an instructive exercise to infer the biosynthetic pathway by retrosynthetic analysis. Given the boundary condition of an indole precursor, the pyridine ring of the quinoline ring system in 337 must be generated. This might be accomplished by dehydrogenation of the imine produced from an amino aldehyde precursor 338. The alcohol functionality in 338 could be the residue of electrophilic activating functionality in a precursor that was involved in a connection with the nucleophilic amino group in a pyrrole ring, as in the tryptophan 339. Referring to the boundary condition of tryptophan as the biosynthetic starting material, the aldehyde in the precursor 339 could be generated by hydrolytic cleavage of an imine derivative 340 of the ethylamine sidechain of tryptamine. A concomitant disconnection of one bond to the tertiary amino group in 339 is required to make room for the connection. Referring to the boundary condition of vincoside as starting material, 340 could arise from acarboxy dialdehyde 341 by polar decarboxylation and heterocyclization.
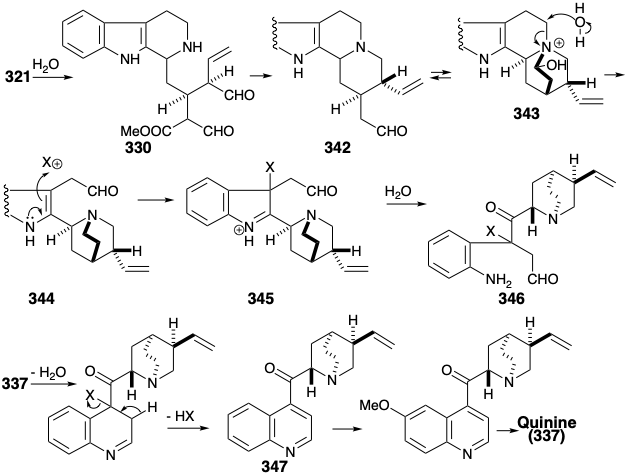
The biosynthesis of the quinuclidine portion of quinine (337) from the secologanin portion of vincoside (321) involves hydrolysis of the glucoside to give a dialdehyde 330, followed by intramolecular reductive alkylation and decarbomethyoxylation to give 342, that is in equilibirum with a hemiaminal 343. Hydrolytic fragmentation of the latter accompanied by oxidation of a primary alcohol to an aldehyde and reduction of the hemiaminal to an amine affords 344. The rearrangement of the indole portion of 344 to a quinoline skeleton is initiated by an oxidation to 345 followed by ring cleaving hydrolysis and recyclization of the resulting amino aldehyde 346. Arene oxidation, methylation, and then reduction of the resulting quinoline derivative 347 delivers quinine (337).
A Relay Synthesis of Quinine
A major topological simplification of the quinine skeleton arises by disconnection of a bond between atoms 1 and 8. Atom 1 is a common atom of the multicyclic quinuclidine portion of 337. Though atom 8 is a noncommon atom, its role as a link between the two major portions of 337 recommends removal of skeletal connections to this atom. This disconnection was actually achieved by Rabe during degradative studies on the structure of 337.17 The fragmentation of 337 to 348 depends upon the polar activation provided by the quinoline and quinuclidine amino groups (ignoring the activation provided by the C-8 hydroxyl).
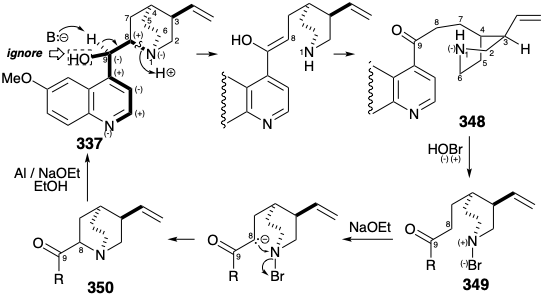
Polar Redox Reactions
Rabe also demonstrated that the reverse process, a synthesis of 331 from 347, can be achieved by exploiting the polar activation of C-8 in 347 (quinine numbering).17 This approach requires a nitrogen electrophile and involves oxidation via polar intermediates. Thus, the nucleophilic secondary amino group in 348 is converted into an electrophile in 349 by appending a more electronegative atom, i.e. bromine. This constitutes oxidation of the amino group. Electrophilic attack on carbon in 349 to give 350 produces a bond between carbon and a more electronegative atom (i.e. nitrogen). This constitutes oxidation of carbon coupled with reduction of the amino group. We shall refer to such reactions as polar redox reactions. An example of this reaction type occurs in the biosynthesis of acetyl CoA (see section 2.3) during nucleophilic attack by hydroxyethylidene TPP (351) on the the disulfide 352. Thus, the nucleophilic carbon is oxidized from f = 1 in 351 to f = 2 in 353 while a sulfur atom in 352 is reduced from f = -1 to f = -2 in 353. This reaction is complex because another carbon in 351 is concurrently oxidized from f = 2 to f = 3 in 353 in conjunction with reduction of the second sulfur in 352 from f = -1 to f = -2 in 353.
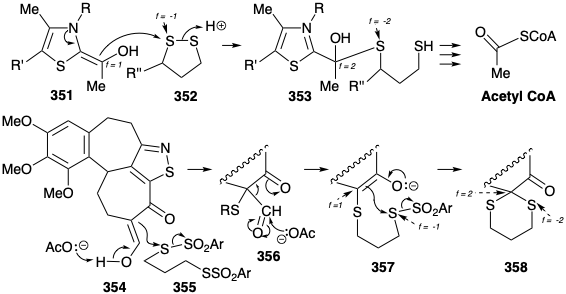
Another polar redox reaction, again with sulfur as electrophile, was encountered in the Woodward synthesis of colchicine (see section 6.1). In fact, the introduction of a dithioketal at the nucleophilic carbon α to a cabonyl group involves two successive oxidations of the α carbon, first of 354 to 356 then of the latter into 358, coupled with two reductions of sulfur, first in the conversion of 355 to 356 then in the conversion of the latter, via 357, into 358. This reaction is also complex because another carbon in 354, α to the enolic hydroxyl, is concurrently oxidized from f = 1 to f = 2 in 356 in conjunction with reduction of a second sulfur in 355, and a second carbon is oxidized from f = -1 in 357 to f = -2 in 358 (the carbonyl carbon) in conjunction with reduction of a second sulfur.
A Convergent Strategy for Key Intermediate 348
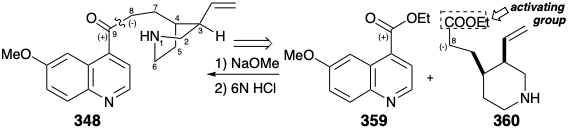
It should be noted that 350 (see above) is a mixture of epimers at C-8, and the reduction that produces 337 introduces another asymmetric center (at C-9). Fortunately, 337 was a major component of the isomeric mixture produced by this nonstereocontrolled conversion of 348 to 337. This conversion makes quinotoxine (348) an attractive subtarget for the total synthesis of quinine (337). The subtarget 348 is further simplified by a dislocation, that breaks the molecule into two large fragments by severing one of the four bonds connecting the quinoline and piperidine rings. The dislocation chosen by Woodward and Doering for the first total synthesis of quinine (337) was dictated by the fact that the reverse process, synthesis of 348 from 359 and 360, had excellent precedent. A dihydro derivative of 348 (with an ethyl instead of a vinyl group) was prepared by Rabe from 359 and a dihydro derivative of 360 which had been obtained from degradation of natural quinine (337).
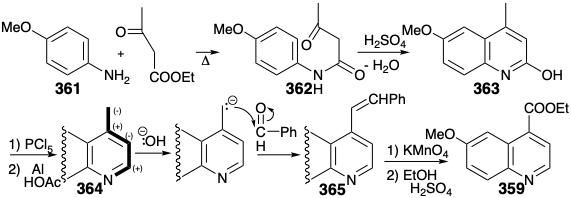
Total syntheses of the subtarget ethyl quininate (359) were also known when the total synthesis of 337 was undertaken. A particularly effective route introduces the carboxyl group in latent form as a benzylic methyl group, and constructs the nitrogen heterocycle on a preformed aromatic precursor 361 by polar reactions. Cyclodehydration of 362 affords 363, that is reduced to 364. Benzylic oxidation of 364 is achieved by oxidative cleavage of a latent carboxylic acid, a C=C bond in the precursor 365, that is available by a polar condensation of 364 with benzaldehyde. The condensation exploits the nucleophilic activation of the benzylic methyl, that is provided by the nitrogen in 364.
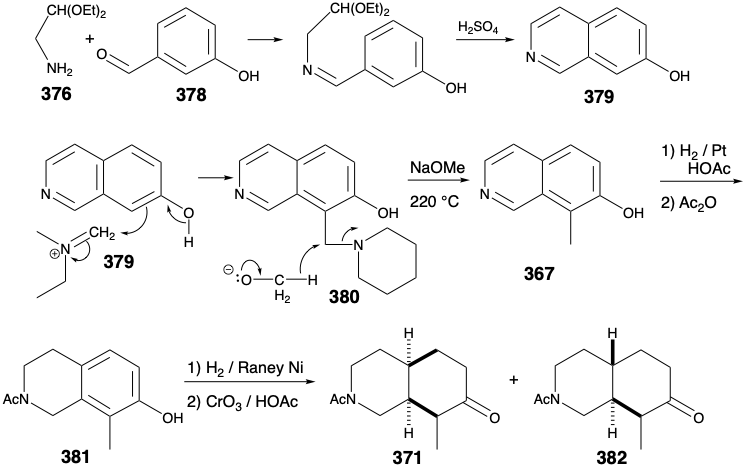
The only remaining synthetic objective was, thus, ethyl N-benzoylhomomeroquininate (360). The two side chains in 360 could be generated stereospecifically cis by oxidative cleavage of a temporary bridge in 366. The cis ring fusion in 366 could be produced, in turn, by catalytic hydrogenation of an aromatic isoquinoline precursor 367.
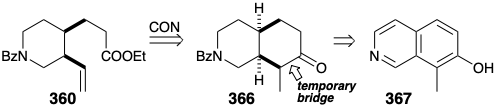
Several potential flaws must be avoided in designing a detailed scheme for the 366 to 360 conversion. For example, cleavage of the temporary bridge in 366 by a Baeyer-Villiger oxidation followed by an elimination to generate the vinyl group in 360 must avoid generating the alternative, thermodynamically favored, ethylidene derivative 368.
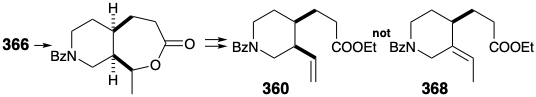
Interestingly, the reaction chosen to achieve the ring cleavage was a reaction used earlier in degradation studies for determining the structure of quinine. Thus, Rabe effected cleavage of quininone (369) into ethylquininate (359) and an oximino compound 370 by treatment with amyl nitrate and sodium ethoxide. This cleavage is analogous to a retro-Claisen reaction, that occurs especially readily for nonenolizable β-keto esters.
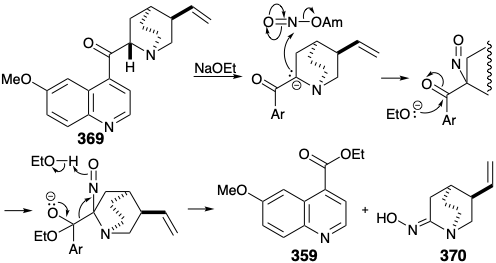
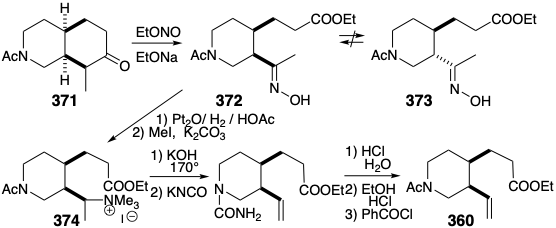
Application of this cleavage process to the N-acetyl analogue 371 of 366 generates oxime 372. A potential flaw, epimerization of the cis-1,2-disubstituted product 372 into the thermodynamically more stable trans isomer 373, was not a problem. In contrast to what would be expected for the corresponding acetyl derivative, the oxime 372 is not prone toward epimerization. Thus, the hydroxyl proton rather than an α-proton is preferentially abstracted upon treatment of oximes with base. Reduction and methylation of 372 readily affords a quaternary ammonium derivative 374, that affords the requisite terminal vinyl group in 360 by base promoted Hofmann elimination involving regioselective abstraction of hydrogen from the less substituted β carbon.
As noted above, an aromatic precursor 367 was chosen for the synthesis of ethyl N- benzoylhomomeroquininate (360). A monocyclic aromatic precursor for 367 could be either a pyridine derivative or a benzene derivative. Choosing the latter allows exploitation of electrophilic aromatic substitution on an electron-rich precursor to accomplish annelation of the pyridine ring. This annelation requires carbon-carbon bonds meta and para to the hydroxyl group. Formation of the para bond by electrophilic aromatic substitution is favored over meta by the strong electron donating activating effect of the hydroxyl group. Formation of this para carbon-carbon bond in the last step of annelation suggests a phenolic precursor 375 with the four atoms of the incipient pyridine ring appended to the meta position. The bond between this meta substituent and the phenol ring can not be generated by electrophilic aromatic subsitution because ortho and para rather than meta substitution is favored. However, disconnection of this substituent by removal of the bond between nitrogen and the benzylic carbon suggests a dissonant carbonyl-masked amino acetaldehyde 376 and a benzaldehyde derivative 377. The methyl substituent in 377 might also be introduced by electrophilic aromatic substitution on the readily available m-hydroxy-benzaldehyde (378). However, achieving the requisite regiocontrol in such an alkylation might be difficult.
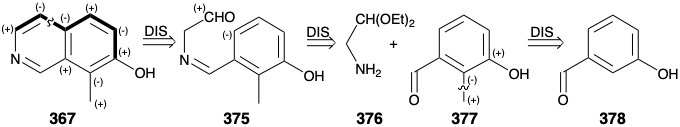
In fact, a different order of steps was adopted. Introduction of the ortho methyl substituent was postponed until after annelation of the pyridine ring was completed because introduction of a methyl group can be readily achieved regioselectively by electrophilic aromatic substitution on the β-hydroxy isoquinoline 379. Thus, aminomethylation with piperidine and formaldehyde produced the benzylic amine 380 that was reduced to 367 upon heating in the presence of sodium methoxide. This unusual reduction involves hydride transfer from methoxide. Technical difficulties arose in the hydrogenation of 367 to 371. Thus, because the amine poisoned the catalyst, the hydrogenation stopped after only the nitrogen containing ring had been reduced. The amine had to be blocked as an amide before reduction of the benzene ring could be achieved. The desired cis-stereospecificity in the reduction of 381 to 371 could not be achieved. Fortunately, however, this was not a fatal flaw because the required isomer could be isolated from the trans-fused hydrogenation reaction product 383. Thus, catalytic hydrogenation is not entirely reliable for stereoselective delivery hydrogen to one face of an aromatic ring.