
6.6: Biosynthesis of Nonaromatic Amino Acids
- Page ID
- 285465

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\) The availability of an appropriate starting material dictates a biosynthetic strategy for alanine (383). Pyruvic acid (384) is a key intermediate in the biosynthesis of acetyl CoA. A functional group interchange (FGI) coupled with functionality level adjustment (FLA), i. e. reductive amination, can provide the α amino substituent of 383 from the carbonyl group in pyruvic acid.
The availability of an appropriate starting material dictates a biosynthetic strategy for alanine (383). Pyruvic acid (384) is a key intermediate in the biosynthesis of acetyl CoA. A functional group interchange (FGI) coupled with functionality level adjustment (FLA), i. e. reductive amination, can provide the α amino substituent of 383 from the carbonyl group in pyruvic acid.
Adopting a similar strategy for the biosynthesis of aspartic acid (385) suggests oxaloacetic acid (386) as a precursor. The consonant circuit in the β-keto acid array of 386 suggests a polar synthesis from pyruvic acid and carbon dioxide.
In the biosynthesis of 386, carboxylation does not involve the direct reaction of pyruvic acid with \(\ce{CO2}\). Rather, the carboxylating agent is an enzyme-bound N-carboxy biotin derivative that is generated by a series of reactions that begin with the activation of carbonate by phosphorylation with ATP. The resulting carbonic phosphoric anhydride acylates the biotinyl nitrogen of N-carboxybiotin which is bound to an enzyme, pyruvate carboxylase, as N-carboxybiocytin. Pyruvate carboxylase catalyzes the transfer of a carboxy group to pyruvate from N-carboxybiotin. Alternatively, in some plant cells phosphoenol pyruvate is carboxylated producing oxaloacetic acid directly.


The biosynthetic strategy for glutamic acid (387) is more intricate. Thus, the potential precursor α-ketoglutaric acid (388) contains dissonant circuits in both the g and the α-keto acid arrays precluding a direct polar synthesis by a C-C connective route from smaller precursors. One way to invert the polar reactivity pattern generated by a functional group is 1,2-transposition of the functionality. Thus, the transposed precursor 389 has consonant β-hydroxy acid arrays that could be created by polar condensation of an acetate C-nucleophile with malonaldehydic acid as a carbonyl electrophile.
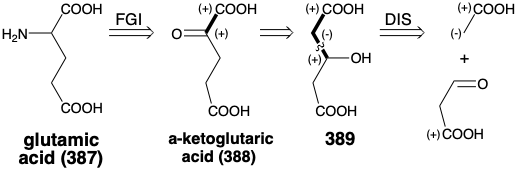
This polar strategy is adopted by Nature except that oxaloacetic acid (386) rather than malonaldehydic acid is used as the electrophile. This starting material has an extra carboxyl group that must be removed during the construction of 388. The 1,2-oxygen transposition also creates a polar pathway for the requisite decarboxylation. The biosynthetic strategy also has other idiosyncrasies. Thus, besides a plan for assembling the carbon skeleton, the biosynthetic strategy for glutamic acid includes cogeneration of a reducing agent. We encountered a similar tactic in the biosynthesis of fatty acids from glucose (see section 3.1) where the conversion of glucose to the starting material, acetyl CoA, cogenerates all the reducing agent, NADPH, required for deoxygenation of β-ketoacyl intermediates. Nature's remarkable strategy for the biosynthesis of glutamic acid simultaneously generates a starting material, α-ketoglutaric acid 388, and the requisite reducing agent, NADPH, for the subsequent reductive amination of 388 to produce 387.

The biosynthesis begins with condensation of an acetyl CoA nucleophile with the highly electophilic carbonyl carbon of oxaloacetic acid to form citric acid (390). The reaction is catalyzed by the enzyme, citrate synthetase (or condensing enzyme). Dehydration of citric acid gives cis-aconitic acid (391) that is then hydrated to isocitric acid (392). Dehydrogenation of the latter is coupled with decarboxylation of a presumed β-ketoacid intermediate 393 to yield α-ketoglutaric acid (388). The hydrogen is transferred to NADP+ generating the NADPH needed for the nitrogen-fixing reductive amination of 388. Thus, the protonated imine 394, that is produced by reaction of the ketone carbonyl with \(\ce{NH3}\), is reduced by hydride transfer from NADPH to deliver L-glutamic acid (387) with enzyme-induced enantioselectivity. This is an example of asymmetric induction by a homochiral reagent (the enzyme) during the reaction of a prochiral intermediate, the imine 394.
Interestingly, an oxidative pathway exists in Nature for conversion of α-ketoglutaric acid (388) back into oxaloacetic acid (386).  Thus, oxidative decarboxylation of α-ketoglutaric acid occurs by the same thiamine pyrophosphate-catalyzed mechanism as for the pyruvate-acetate conversion (see section 2.3) that cogenerates NADH from NAD+. The initial product, succinyl CoA (385), is a high energy thioester. Its hydrolysis is coupled with phosphorylation of ADP, through an indirect process involving phosphorylation of a histidine residue of the hydrolyzing enzyme (see 399), transfer of phosphate to guanosyl diphosphate (390, GDP), and finally, transfer of phosphate from the resulting GTP to ADP. Dehydrogenation of succinic acid (396) is then catalyzed by succinate dehydrogenase producing fumaric acid (397). The hydrogen is transferred to flavin adenine dinucleotide (401, FAD) producing FADH2. The reversible hydration of fumaric acid to give L-malic acid (398) is catalyzed by fumarase, that promotes enantioselective stereospecifically trans addition of water to the symmetrical prochiral olefin. This is another example of asymmetric induction by a homochiral reagent, the enzyme fumarase. Finally, L-malate dehydrogenase catalyzes the oxidation of L-malic acid to oxaloacetic acid by transfer of hydride to NAD+.
Thus, oxidative decarboxylation of α-ketoglutaric acid occurs by the same thiamine pyrophosphate-catalyzed mechanism as for the pyruvate-acetate conversion (see section 2.3) that cogenerates NADH from NAD+. The initial product, succinyl CoA (385), is a high energy thioester. Its hydrolysis is coupled with phosphorylation of ADP, through an indirect process involving phosphorylation of a histidine residue of the hydrolyzing enzyme (see 399), transfer of phosphate to guanosyl diphosphate (390, GDP), and finally, transfer of phosphate from the resulting GTP to ADP. Dehydrogenation of succinic acid (396) is then catalyzed by succinate dehydrogenase producing fumaric acid (397). The hydrogen is transferred to flavin adenine dinucleotide (401, FAD) producing FADH2. The reversible hydration of fumaric acid to give L-malic acid (398) is catalyzed by fumarase, that promotes enantioselective stereospecifically trans addition of water to the symmetrical prochiral olefin. This is another example of asymmetric induction by a homochiral reagent, the enzyme fumarase. Finally, L-malate dehydrogenase catalyzes the oxidation of L-malic acid to oxaloacetic acid by transfer of hydride to NAD+.

The overall process, generation of α-ketoglutaric acid from acetyl CoA plus oxaloacetic acid and regeneration of oxaloacetic acid from α-ketoglutaric acid also produces two molecules of \(\ce{CO2}\), four molecules of reducing agent (2 x NADH, NADPH, and FADH2), and one molecule of ATP. This cycle of reactions, known as the Krebs cycle or the tricarboxylic acid cycle (citric acid is a tricarboxylic acid) results in the aerobic oxidative catabolism of acetyl CoA. Besides providing a source of useful reagents for biosynthesis from fatty acids or sugars (via acetyl CoA), it also generates a variety of biosynthetically useful intermediates, e.g., α-keto glutaric acid) that can be diverted from the cycle. If Krebs cycle intermediates are to be removed from the cycle for biosynthesis, then other cycle intermediates must be generated somehow to replace them. The most important anaplerotic (filling up) reaction is the pyruvate carboxylase-catalyzed carboxylation of pyruvate to form oxaloacetate. Another modification of the Krebs cycle, known as the glyoxylate cycle, is important in plants and microorganisms for production of biosynthetic starting materials from acetyl CoA. Acetyl CoA condenses with oxaloacetic acid giving isocitric acid by way of citric and cis aconitic acids. But, rather than being oxidized to α-ketoglutaric acid, isocitric acid is cleaved to succinic and glyoxalic acids in a retro-aldol reaction that is catalyzed by isocitrase. Succinic acid may then be used for biosynthesis, while glyoxalic acid (392) reenters the Krebs cycle by malate synthetase-catalyzed condensation with aceyl CoA to form L-malic acid.

Glutamic acid is the direct biosynthetic precursor of glutamine (404) and proline (406). Activation of one carboxyl as a mixed phosphoric carboxylic anhydride (403) followed by acylation of \(\ce{NH3}\) delivers 404. Selective partial reduction of one carboxyl and intramolecular reductive amination of the resulting γ-amino aldehyde 405 delivers 406.

The biosynthesis of glutamic acid from α-ketoglutaric acid by reductive amination with ammonia is not typical. Generally, the conversion of α-keto acids into the corresponding amino acids involves transfer of an amino group from glutamic acid, a process called transamination. Pyridoxal phosphate (407) and divalent metal cations are cocatalysts for the transfer that occurs by a polar process. Thus, the imine from 4 0 7 and glutamic acid undergoes prototropic shift to generate the tautomer 4 0 9 . Rearomatization by another prototropic shift generates a new imine 410 that is hydrolyzed to pyridoxamine phosphate (411) and α-keto glutaric acid. Transamination is then completed by the reaction of an α-keto acid with 411 to produce the corresponding α-amino acid and regenerate pyridoxal by a process that is analogous to the reverse of the reaction that generates 411 plus α-keto glutarate from 407 plus glutamic acid.

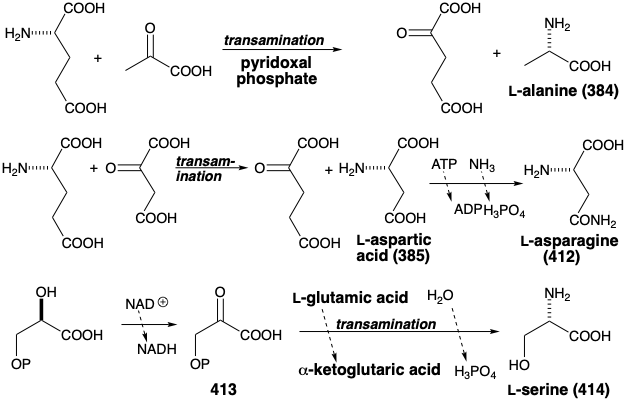
Thus, transamination of pyruvic and oxaloacetic acids yields alanine (384) and aspartic acid (385), respectively. The biosynthesis of asparagine (412) from 385 parallels that of glutamine from glutamic acid (see above). Serine (414) is produced in Nature from 3-phosphoglyceric acid via oxidation to 3-phosphopyruvic acid (413), followed by transamination with glutamic acid.
The consonant circuit between the carboxyl and hydroxyl groups of serine (414) suggests a polar strategy for the biosynthesis of glycine (415) involving retroaldol cleavage. The α-amino group is in a dissonant relationship with the other functional groups in 415 and, therefore, cannot assist in the polar cleavage. However, Nature adopts a strategy that allows the desired cleavage to occur under mild conditions by prior umpölung of the α-amino group as in 416 that has two functional groups stabilizing the buildup of electron density at the α-carbon.

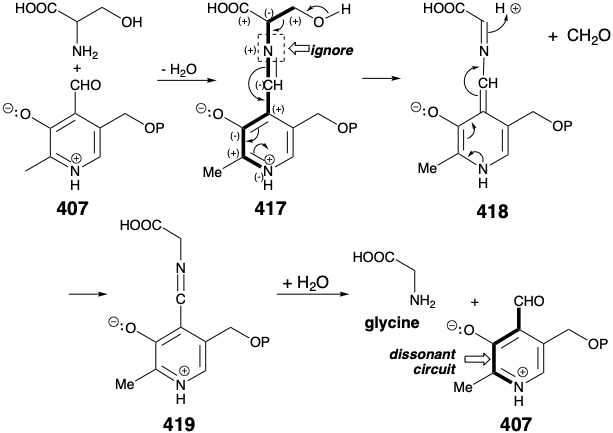
Pyridoxal phosphate (407) is the reagent, a polar reactivity inversion operator, that converts the amino group by a polar process into a derivative in 417 which stabilizes an anion at the α-carbon. It is instructive to consider how this process works. The key feature of the reagent 417 is a dissonant relationship between the aldehyde and pyridinium nitrogen. The electrophilic reactivity of the aldehyde carbon is used to form a C=N bond with the nucleophilic amino nitrogen in serine. The polar reactivity of this C=N bond is then ignored, and it is the dissonant pyridinium nitrogen in 417 that stabilizes the buildup of electron density on the serine α-carbon. The C=N bond derived from the serine amino group serves only to conjugate the pyridinium nitrogen with the serine α-carbon. Retroaldol fragmentation of 417 produces 418 and formaldehyde. The latter is captured by tetrahydrofolic acid (vide infra) while 418 is rearomatized and protonated to produce an imine 419 of glycine. Hydrolysis produces glycine and regenerates pyridoxal phosphate that is, thus, a true catalyst for the retroaldol fragmentation.
The formyl group lost in the conversion of 417 to 418 is transferred to tetrahydrofolic acid (420, FH4). The product, N5,N10-methylene FH4 (421), is one member of a family of folic acid coenzymes that carry one-carbon groups, such as methyl, formimino, and formyl in 422, 423, and 424, respectively.

The major portion of these one-carbon transfers is achieved via methyl transfer to homocysteine (425) from N5-methyltetrahydrofolate (422), which yields methionine (426) and, hence, S-adenosylmethionine (SAM+). Hence, serine and, ultimately, 3-phosphoglyceric acid is the source of the ubiquitous methyl groups donated by SAM+ to a wide variety of acceptors.

One biosynthetic strategy for L-lysine (427) is closely related to that involved in the biosynthesis of glutamic acid (see above). Thus, double reductive amination of a precursor α-keto diacid 428 could provide the two amino groups in 427. A precursor 429 containing a consonant β-hydroxy acid array is suggested by 1,2-transposition of oxygen functionality in 428. The precursor 429 could be created by polar condensation of an acetate C-nucleophile with succinaldehydic acid as a carbonyl electrophile.
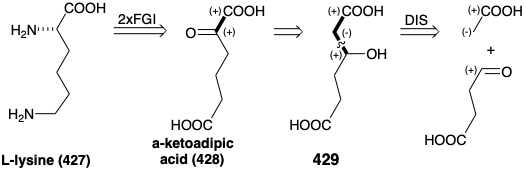
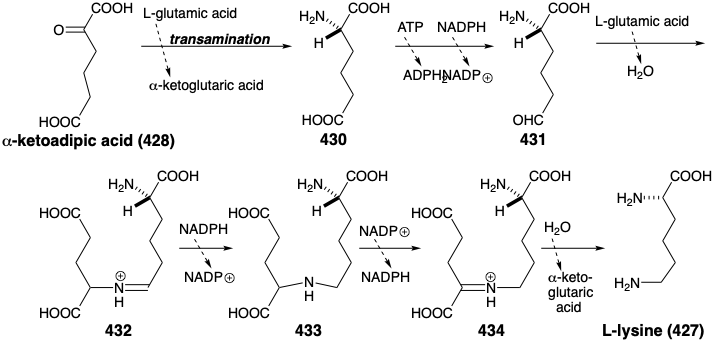
This is the biosynthetic strategy for lysine that is adopted by most fungi except that α- ketoglutaric acid (388) rather than succinaldehydic acid is used as the electrophile. The biosynthesis proceeds via α-aminoadipic acid (430) by a scheme commencing with a Claisen-Schmidt condensation between α-ketoglutaric acid (388) and acetyl CoA. Rearrangement, oxidation, and decarboxylation occur in reactions analogous to the conversion of oxaloacetic acid into α-ketoglutaric acid (see above). Transamination of the resulting a-ketoadipic acid (428) enantioselectively produces the L isomer of α-aminoadipic acid (430). Reduction of 430 to 431 followed by reductive alkylation of glutamic acid by 431 affords 433. Oxidation followed by hydrolysis gives L-lysine (427). Note that the iminium group in 434 is stabilized relative to that in 432 owing to conjugation with the carboxyl in 434.

An alternative strategy for the biosynthesis of L-lysine (427) generates the ε-amino array by decarboxylation of an α-amino acid group in a symmetrical precursor 435, a process which can be catalyzed by pyridoxal phosphate (see below). As is common in α-amino acid biosynthesis, 435 could be derived from an α-keto acid precursor 436. The carbon skeleton of this ketone can be assembled by an aldol condensation between a pyruvic acid enolate and an aldehyde electrophile 437 derived from L-aspartic acid (385).

The primary route for biosynthesis of L-lysine in bacteria and higher plants generates a seven carbon diacid intermediate 438 from the three carbons of pyruvic acid and four carbons of aspartic acid (385). Reduction of 385 to the aldehyde 437 with concomitant hydrolysis of ATP is analogous to the reduction of 3-phosphoglyceric acid (3PG) to glyceraldehyde-3-phosphate (G3P) (see section 2.1) and the production of 431 from 430. Aldol condensation of 437 with pyruvic acid delivers 438. Intramolecular imine formation and dehydration produces 439 that is reduced and then hydrolyzed and succinoylated to produce a masked (N-succinylated) derivative 440 of 2-amino-6-ketopimelic acid. Transamination and hydrolysis of the resulting amino amide 441 delivers diamine 435. Monodecarboxylation of this α-amino acid, catalyzed by pyridoxal phosphate, then delivers L-lysine (427). As in the pyridoxal phosphate-catalyzed retro-aldol cleavage of serine to glycine (see above), pyridoxal serves as a polar reactivity inversion operator that temporarily converts the amino group by a polar process into a derivative in 442 which stabilizes electronic excess at the α-carbon. The consonant circuit between the pyridinium nitrogen and carboxyl carbon in 442 (ignoring the polar reactivity of the imine group) facilitates polar cleavage of a C-C bond generating the imine 443. Aromatization by prototropic shift then produces 444. Finally, the polar reactivity of the imine is utilized to achieve hydrolysis, releasing the amine 427 and regenerating the aldehyde catalyst 407.

Note that both the α-amino acid array in 435 and the amino aldehyde array in 407 incorporate dissonant circuits. It is the polar union of one functional group in each dissonant reactant (407 and 435) that creates a consonant relationship between the remaining functional groups in these molecules. Thus, umpölung of the amino group in α-amino acids is accomplished by a dissonant difunctional reagent, pyridoxal pyrophosphate. Previously umpölung of the ketone carbonyl group in α-keto acids was encountered in the cyanide ion catalyzed benzoin condensation (section 2.1), in the thiamine pyrophosphate (TPP) catalyzed transketolase reaction (section 2.1), and in the TPP catalyzed decarboxylation of pyruvic acid (section 2.2). It is now instructive to note that both HC≡N and TPP contain biphilic functionality. They are both readily metallated (i.e. deprotonated). This introduces a nucleophilic functional group (the carbanion) in a dissonant relationship with other functionality in these molecules, and allows them to serve as catalytic polar reactivity inversion operators. One of the functional groups, the nucleophilic carbanion, is exploited to link the catalyst to the ketol or aldehyde carbonyl, in the transketolase or benzoin reactions respectively, or the α-keto acid carbonyl in the pyruvate decarboxylation reaction. The other functionality in the catalyst then stabilizes electronic excess at the formerly electrophilic ketone or aldehyde carbonyl carbon.