
6.4: Lysergic Acid
- Page ID
- 285468

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis of Tryptophan and Lysergic Acid
The carbon skeleton of anthranilic acid (254) is constructed in nature from two molecules of phosphoenol pyruvate and one of erythrose 4-phosphate via chorismic acid (see section 5.1). The two-component enzyme complex of anthranilate synthetase (AS) then promotes the transfer of \(\ce{NH3}\) from glutamine to chorismic acid (4) affording amino acid 253 by conjugate displacement of a hydroxyl group. During the strategically intricate biosynthesis of tryptophan (259), the carboxyl carbon of 254 is ultimately lost. A five-carbon unit from ribose is appended to the amino group of 254, but suprisingly all five carbons of phosphoribosyl pyrophosphate (PRPP) are not retained to complete the tryptophan skeleton (vide infra). Deprotonation of the imine derived from 255 delivers an enamine 256 that is also the enol tautomer of ketone 257. Intramolecular Friedel-Crafts cyclization of the latter delivers a β-iminocarboxylic acid 258 that, being the nitrogen analogue of a β-keto acid, readily decarboxylates producing indole-3-glycerol phosphate. While the biosynthesis of tryptophan might be completed by simple functionality adjustments, Nature adopts a different, more convoluted, strategy. Thus, tryptophan synthetase catalyzes a remarkable Friedel-Crafts alkylation with L-serine coupled with a dealkylation that cleaves glyceraldehyde-3-phosphate (G3P) and delivers L-tryptophan (259).

So far, we have seen that complex alkaloids may be assembled in Nature by a convergent strategy involving the union of two large fragments derived from aromatic amino acids. Alkaloids may also be constructed in Nature by the conjugation of amino acid-derived intermediates with terpenoid starting materials. Thus, as outlined below, lysergic acid (270) is forged from tryptophan (259) and Δ2-isopentenyl pyrophosphate. Later, we shall see how indole 259 is united in a variety of ways with secologanin, a terpene, to generate a vast array of structurally complex tryptophan-derived alkaloids. The carbons of these starting materials remain connected in the product, although the carboxyl carbon is lost. Thus, the biosynthetic strategy is simple.

The biosynthesis of lysergic acid commences with the prenylation of tryptophan. Thus, Δ2-isopentenyl pyrophosphate is a potent electrophile that readily alkylates the nucleophilic benzene ring of tryptophan 259 to afford 4-prenyltryptophan (260). The cyclization of 260 to lysergic acid requires addition of functionality to the Δ2-isopentenyl (prenyl) group by oxidations. The process is accompanied by a remarkable odyssey of the allylic carbon marked with an asterisk in the intermediates 260 - 265. Allylic hydroxylation and dehydration provide 261. The diene 261 has free rotation, that allows interconversion of the E and Z-methyl carbons during the 260 to 263 conversion. Formation of the D-ring is believed to occur by a decarboxylative SN2' alkylation in the allylic epoxide 262. Oxidation of the cyclization product, chanoclavine-I (263), to an E allylic aldehyde 264 is followed by cis-trans isomerization to the Z-isomer 265. Condensation to the Schiff base 266 completes the lysergic acid skeleton. Final adjustment of functionality by reduction to agroclavine (267), allylic oxidation to elymoclavine (268), further oxidation to Δ8-lysergic acid (269), and isomerization gives lysergic acid (270).
Dihydrogen as a Masking Group for an Alkene
Lysergic acid is thermodynamically unstable. Acids, base, or noble metals readily catalyze the irreversible rearrangement of lysergic acid (270) into a naphthalene isomer 271 by migration of a D-ring and a B-ring C=C bond into the C ring. Therefore, the stability of aromatic derivatives, that may often be advantageously exploited in complex molecular synthesis, was seen by Woodward as a major obstacle for the synthesis of lysergic acid (270). A central tactic in the first successful synthetic strategy,13 was the scrupulous avoidance of aromaticity in ring C. On the other hand, the crucial last step of the scheme, dehydrogenation of an indoline 272, ingeniously exploits the aromaticity of the indole array in 270. What is remarkable about this strategy is Woodward's recognition that, although it might seem unlikely that a way could be found to dehydrogenate 272 without also inducing isomerization of 270 to 271, the search for a method to achieve such a selective reaction could provide an excellent solution to the central challenge of the synthesis, and, therefore, was well worth the effort.
into a naphthalene isomer 271 by migration of a D-ring and a B-ring C=C bond into the C ring. Therefore, the stability of aromatic derivatives, that may often be advantageously exploited in complex molecular synthesis, was seen by Woodward as a major obstacle for the synthesis of lysergic acid (270). A central tactic in the first successful synthetic strategy,13 was the scrupulous avoidance of aromaticity in ring C. On the other hand, the crucial last step of the scheme, dehydrogenation of an indoline 272, ingeniously exploits the aromaticity of the indole array in 270. What is remarkable about this strategy is Woodward's recognition that, although it might seem unlikely that a way could be found to dehydrogenate 272 without also inducing isomerization of 270 to 271, the search for a method to achieve such a selective reaction could provide an excellent solution to the central challenge of the synthesis, and, therefore, was well worth the effort.

The Woodward strategy was channeled by the decision to use an intramolecular Friedel-Crafts acylation of the electron-rich A ring in 274 to generate the C ring in 273. This approach is recommended by the ready availability of 274 as starting material, and by the potential utility of the carbonyl group in 273 to activate bond forming reactions required to add the D ring. However, construction of 272 from 273 can be achieved by polar reactions only if polar reactivity inversions are employed, because the polar reactivity patterns of 272 and 273 are opposed.
The ABC-ring intermediate 273 is readily available from 275 by selective catalytic hydrogenation followed by intramolecular Friedel-Crafts acylation of 274. The carbonyl group in 273 provides activation at the adjacent methylene position that may be exploited for the attachment of the requisite nitrogen atom. However, an amino group is a nucleophile. To allow polar C-N bond formation, the potential nucleophilic reactivity of the methylene α to a ketone carbonyl must be inverted. This was achieved by bromination to afford 276 in excellent yield. An early attempt at alkylation of the amine 277 with 276 was unsuccessful. After exploring a very large number of alternative approaches for annelation of the D ring and developing an eleven stage sequence for preparing 278 from 273, it was discovered that a nonpolar solvent was uniquely effective for the alkylation of 277 with 276. Under these reaction conditions, the ketone ketal 278 was produced in excellent yield. This scenario is a poignant epitome of the vicissitudes of organic synthesis. It serves to underscore a caveat mentioned earlier (see section 1.2) that is worth repeating: as the availability of starting materials or methods (new or more effective) for uniting and manipulating them vary, so will the relative merits of different pathways. A poor synthesis can become the method of choice if a way to improve a bad step can be discovered.

It is instructive to examine the alternative strategy for the synthesis of 278 from 273, and to keep in mind that this alternative sequence was one of a great many that were painstakingly explored. The alternative route involves a strategy analogous to the 276 → 278 conversion, except that the carbonyl group of 276 and 278 is present in latent form as a vicinal diol in the corresponding key intermediates 280 and 279 respectively. Thus, the carbonyl is generated in the last step of the synthesis by oxidative cleavage.

The synthesis of 279 involves a novel sequence in which the enol 283 from decarboxylation of the glycidic acid 282 is intercepted by bromine (\(\ce{Br2}\) is in equilibrium with \(\ce{Br3^-}\)) delivering α-bromoaldehyde 284 from the Darzens condensation product 281. Dehydrohalogenation of the α-bromoaldehyde 284 was effected by the mild Mattox-Kendall procedure via semicarbazone 285 and unsaturated semicarbazone 287, that afforded the unsaturated aldehyde 288 by transfer of the semicarbazide residue to pyruvic acid. Nucleophilic epoxidation delivered the key intermediate 280. The latent carbonyl in 279 was deblocked by oxidative cleavage with periodate. This cumbersome route to 278 was abandoned when conditions for achieving the direct preparation of 278 from the bromoketone 276 were discovered.

Completion of ring D was straightforward via intramolecular aldol condensation of the diketone 289. The carbonyl group in the resulting enone 290 then provided reactive functionality in an alcohol 291 and the derived chloride for attachment of cyanide (a carboxy carbanion equivalent) as the final carbon atom of lysergic acid. After hydrolysis of 292 to 272, the two hydrogen atoms, placed at the outset by design at C-5 and C-5a to mask a rearrangement-prone C=C bond, needed only to be removed to afford lysergic acid (270).

A Polar D Ring Annelation Exploiting Target-related Functionality
The D-ring ester and amino functionality provide entirely consonant polar reactivity patterns in the derivative 293 of Woodward's carboxylic acid intermediate 272. An alternative strategy14 for annelation of the D-ring of lysergic acid exploits the polar activation provided by these functional groups that suggests a dislocation to 294. Dislocation of 294 to the aldehyde 288, prepared previously by Woodward, suggests an ylide precursor 295 in which a t-butyl ester serves as a latent amine. A more direct approach using an α-amino ylide seemed inadvisable in view of a possible β-elimination.

Introduction of the amino substituent was initiated by selective hydrolysis of the t-butyl ester in 296 under acidic conditions. The carboxylic acid 297 was then transformed into the corresponding chain shortened primary amine 301 in 80% yield by a Curtius degradation. Cyclization, i.e. of 294, accompanied the reductive amination of formaldehyde with primary amine 301 to afford a 3:1 mixture of the desired ester 293 and its C-8 epimer.

C-Ring Annulation by Nucleophilic Aromatic Substitution
Another synthesis of the key ester intermediate 302 involves annelation of ring C in a preformed ABD-ring precursor by intramolecular nucleophilic substitution with a carbanion 303 that is conjugated with the carboxyl functionality found at position 8 in the target.15 The same carboxyl also provides nucleophilic activation at position 4. Thus, polar analysis of 304 suggests a polar dislocation to two aromatic starting materials that can be joined by an aldol condensation between ketone 305 and carbanion 306. However, this strategy is fatally flawed owing to a preference for ketone 305 to exist as an enol. The addition of another carbonyl group at position 2, as in 309, precludes enolization (blocking group), enhances the electrophilicity of the ketone carbonyl, in 309, and activates the C=C bond in 308 toward dissolving metal reduction.

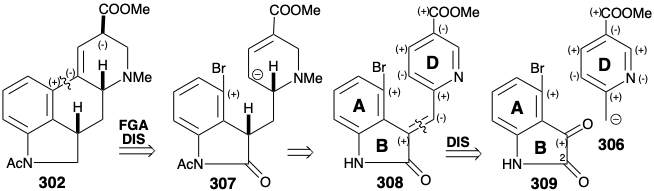
Actually, generation of an isomer of 309, i.e. 310 with the bromo group para to nitrogen, is favored during a synthesis by electrophilic aromatic substitution owing to the powerful activating influence of the nitrogen substituent. However, this is not a flaw because a mecahanism exists for nucleophilic aromatic substitution with rearrangement. Thus, elimination of the nucleofuge leads to a benzyne intermediate 315 to which the nucleophile then adds regioselectively at the required position to give 316. Having served its roles as a reactivity control element, the carbonyl group in 311 is then selectively removed by reduction with diborane. The pyridine ring of the product 312 is activated toward hydride reduction by N-methylation after acetylation of the indole nitrogen. Unfortunately, hydride reduction of the D-ring in 313 produces two epimers only one of which, i.e. 314, cyclizes upon treatment with base delivering 302 in only 15% yield.
