6.3: Morphine
- Page ID
- 285467

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis of Benzylisoquinoline Derived Alkaloids
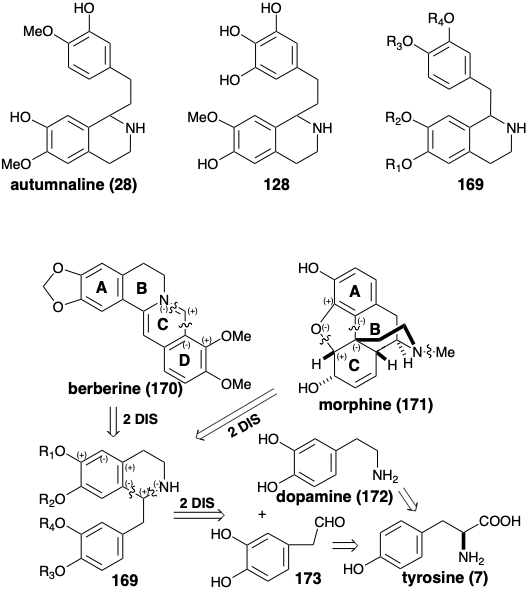
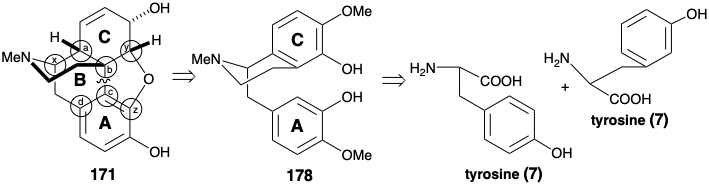
The biosyntheses of colchicine and cephalotaxine involve phenylethylisoquinoline progenitors 28 and 128. Many alkaloids, that are derived biologically from two molecules of tyrosine, share benzylisoquinoline progenitors of general structure 169 (see below). In addition, both six membered rings derived from the aryl nuclei are often retained. Both may be aromatic as in berberine (170), or one may be nonaromatic as in morphine (171). The biogenetic strategy for berberine (170) involves a simple dislocation to a benzylisoquinoline precursor by disconnection of a one-carbon electrophile from the nucleophilic nitrogen and D-ring arene. An intact benzylisoquinoline structure is less evident in the convoluted multicyclic skeleton of morphine (171). If an electron rich aromatic precursor is presumed for the highly oxygenated C-ring, then polar disconection of a furan C-O bond suggests that oxidative coupling of aromatic A and C-rings of a benzylisoquinoline precursor can generate the B-ring of 171. The key benzylisoquinoline intermediates 169 could be produced by Mannich reactions. These condensations might involve polar bond formation between a phenylacetaldehyde electrophile 173 and dopamine (172) as bisnucleophile.
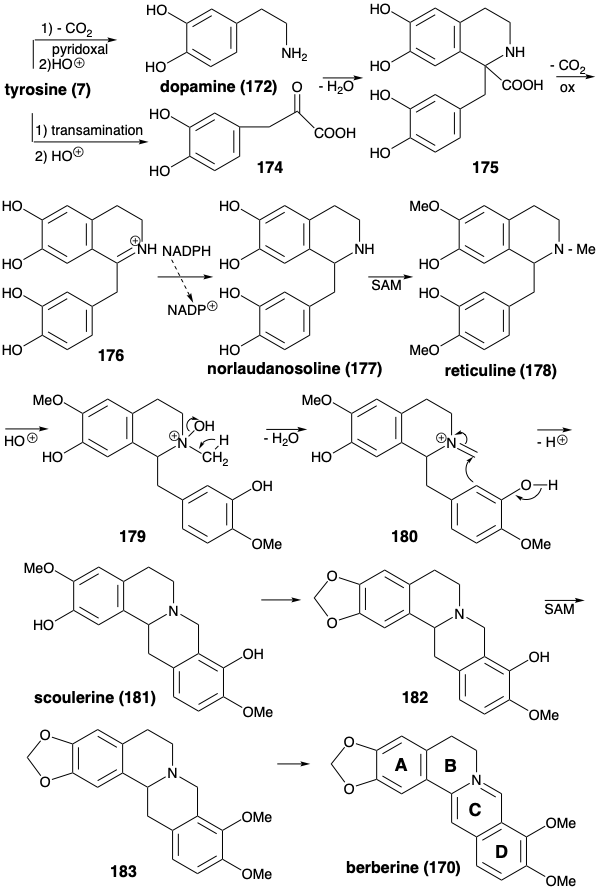
The biosynthesis of berberine (170) from two molecules of tyrosine (see below) commences with pyridoxal-catalyzed decarboxylation and electrophilic hydroxylation that produces dopamine (172). Replacement of the α-amino group of tyrosine with a carbonyl by transamination and electrophilic hydroxylation produces 3,4-dihydroxyphenylpyruvic acid (174). This highly reactive ketone, rather than a phenylacetaldehyde, serves as the electrophile in a Mannich reaction with dopamine (172). Polar condensation of the highly electrophilic carbonyl in 174 with 172 as bisnucleophile generates the benzylisoquinoline ring system in 175. Oxidative decarboxylation of this α-amino acid followed by reduction of the intermediate 176 delivers norlaudanosoline (177) from which reticuline (178) is produced by O and N-methylation. The N-methyl group is incorporated into the berberine skeleton by a Mannich condensation of iminium derivative 180 produced by dehydration of an intermediate protonated N-oxide 179. Conversion of an o-methoxyphenol array in the product 181 to a methylenedioxy group in 182, methylation and aromatization of the product 183 delivers berberine (170).
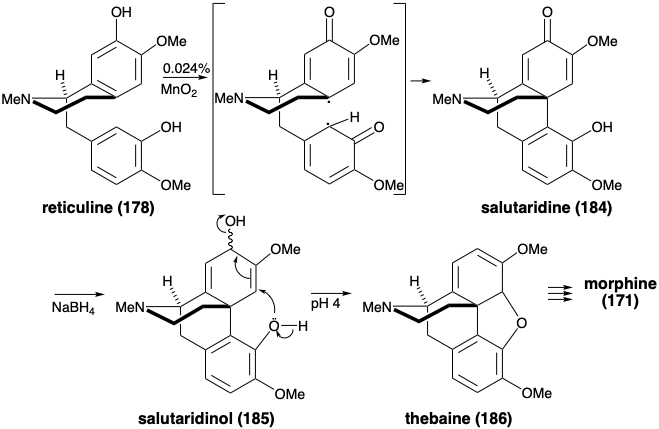
The more topologically complex skeleton of the morphine alkaloids is also produced from reticuline (178). Thus, oxidative ortho-para coupling delivers salutaridine (184). Generation of the benzofuran ring of thebaine (186) occurs after adjustment of functionality level resulting in loss of functionality from position 7 of 185. Interestingly, although simple hydroly-sis of enol ether 186 could produce ketone 188, the oxygen of the methoxyl group is retained in 188. Therefore, a different mechamism must be involved. Perhaps demethylation of 186 occurs through an oxidized intermediate 187 that undergoes retroene fragmentation. Allylic isomerization, reduction, and demethylation then deliver morphine (171).
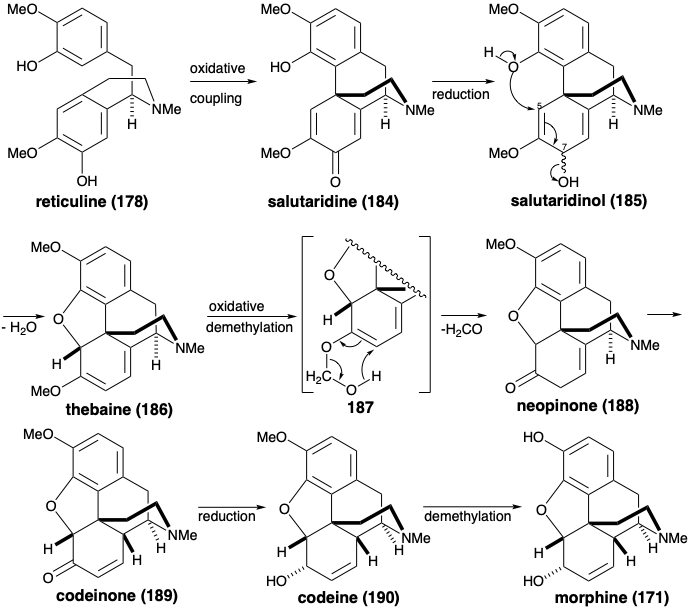
The demethylation of codeine (190) and of the enol ether 187, as well as the conversion of ortho methoxy phenols 137 into methylenedioxy derivatives 139 (see section 6.2) may all be related mechanistically by the initial oxidative conversion of a methyl ether into an α-oxygen-stabilized carbocationic intermediate. Demethylation would occur upon nucleophilic capture by water and fragmentation of the resulting formaldehyde hemiacetal.
A Biomimetic Synthesis of Morphine
Morphine has been assembled in the laboratory by a biomimetic strategy involving oxidative coupling of reticuline (178).9 Oxidative coupling of 178 was accomplished by treatment with manganese dioxide. Salutaridine (184) was obtained, albeit in miniscule yield. Hydride reduction provided the allylic alcohol 185. With mild acid catalysis, 185 underwent intramolecular SN2' displacement of the allylic hydroxyl by the phenolic hydroxyl to afford thebaine (186) from which morphine (171) can be produced (vide infra).
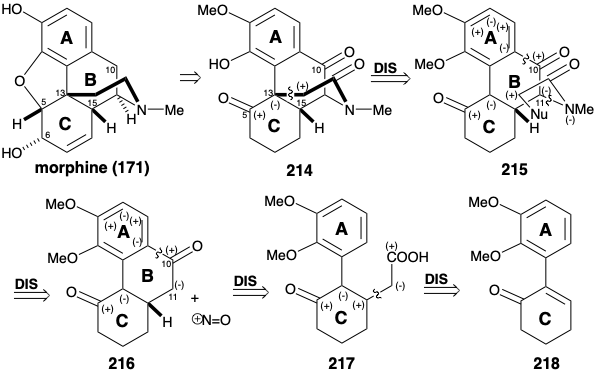
The bridged multicyclic skeleton of morphine has considerable topological complexity. A topological analysis (see section 4.4) may, therefore, be useful for synthetic planning. Considering only the carbocyclic skeleton of 171, there are four common atoms, a-d, and three possible disconnections between them. Of these disconnections, only one, removal of the bond between common atoms b and c, leads to a structural simplification. If the heterocyclic skeleton is also considered, there are also three more common atoms, x, y, and z. Disconnection of bonds between these latter common atoms and a heteroatomic ring member are generally trivial because the heteroatoms are reactive functionality. Disconnection of the b-c bond (and the bond between common atom y and oxygen) suggests a precursor such as 178 (reticuline), the biosynthetic progenitor of morphine alkaloids. Interestingly, this is the only cleavage of a bond between a pair of common atoms that leads to simplification of the morphine carbon skeleton. Thus, cleavage of the bond between common atoms a and b leads to an intermediate with two fused ten membered rings, that would be a redoubtable synthetic challenge. Because it does not lead to reduction of molecular complexity, this dislocation is probably not useful. Cleavage of the π-bond between common atoms c and d disrupts an aromatic system and creates a ten membered ring. The stability of aromatic systems usually disfavors synthetic strategies involving annelation of aromatic rings in the final stages of a synthesis. Therefore, this dislocation is also probably not useful.
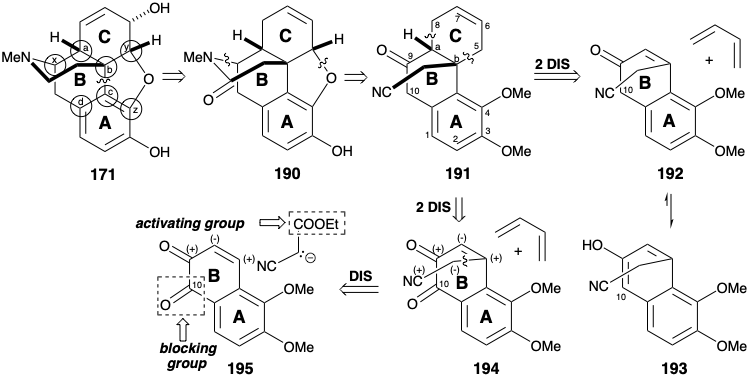
A Diels Alder Strategy for C Ring Annulation
The presence of a cyclohexene array in the C ring of morphine (171) recommends consideration of a Diels-Alder reaction to generate two vicinal exendo bonds, each involving one common atom (i. e. a or b) and one noncommon atom. This topological simplification was exploited in the first total synthesis of morphine.10 However, the C=C bond in the C ring of 171 is in the wrong location. Therefore, with the use of a Diels-Alder tactic as a boundary condition, dislocation of 171 to an amide 190 sets the stage for a retro Diels Alder dislocation. To allow the incorporation of a C=C bond for a dienophile, 190 is first dislocated to 191 by cleavage of carbon-heteroatom bonds to the common atoms x and y. The carbonyl at position 9 in 191 provides activation for a Diels-Alder construction of the C-ring from an AB-ring dienophile 192 and 1,3-butadiene, a relatively electron-rich diene. However, this strategy is fatally flawed because 192 is expected to exist almost exclusively in the aromatic enol form 193 that would not be a reactive dienophile. To block this undesirable enolization, a carbonyl group can be exploited at position 10 (morphine numbering) in a precursor 194. The cyanomethyl sidechain can be appended by the polar union of a nitrile-stabilized nucleophile with the electrophilic β carbon of the α,β-unsaturated carbonyl array in 195. The C-10 carbonyl in 195 also would facilitate this Michael addition of the sidechain nucleophile by preventing enolization of the enone in conjunction with aromatization.
The scheme used for the synthesis of 195 exploits the symmetry of 2,6-dihydroxynaphthalene (196) that readily undergoes electrophilic substitution at the α-position. The electron withdrawing effect of the benzoyl group in the monobenzoate 197 diminishes the electron donating ability of the benzoylated hydroxyl. Therefore, nitrosation occurs regiospecifically at the α-position next to the free hydroxyl. Reduction of the nitroso group affords an amine 198, that is oxidized to an ortho quinone 199. Reduction, methylation, and saponification then delivers phenol 200 that affords 195 by regioselective nitrosation, reductive N-O cleavage and oxidation.
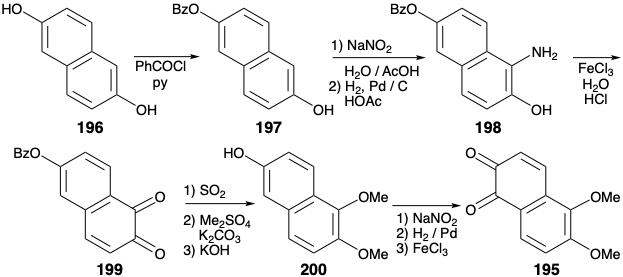
The carbon skeleton of the dienophile 194 is completed by Michael addition of ethyl cyanoacetate carbanion to 195. After saponification, decarboxylation and aromatization, an intermediate hydroquinone 201 is oxidized to give the ortho quinone 194. The carbocyclic skeleton of morphine is completed by a Diels-Alder cycloaddition that provided 191. Elaboration of a piperidine ring began with a reduction that gave the lactam 202 directly. This lactam is epimeric with the morphine skeleton at position 14 presumably owing to steric approach control during delivery of hydrogen to the enolic 9-14 C=C bond in 191.
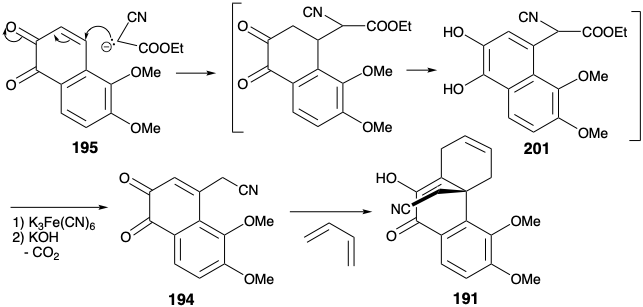
The resulting alcohol presumably adds to the C≡N bond producing an iminoether intermediate that rearranges to the lactam 202. Remarkably, the sterically far more congested 9-14-C=C bond is reduced while the 6,7-C=C bond remains unreduced under these conditions.
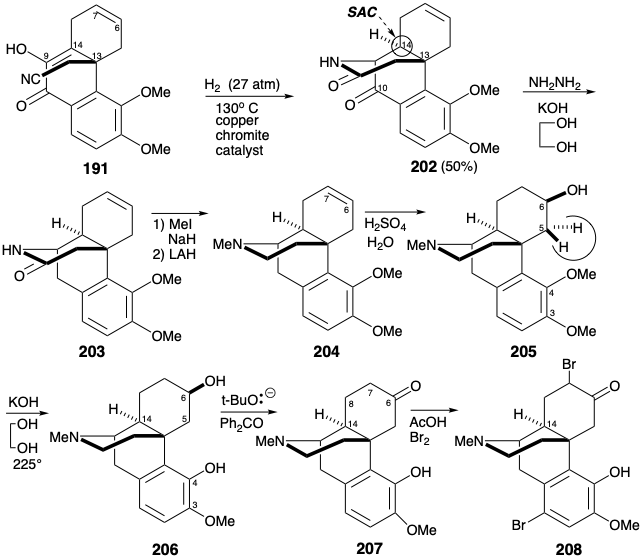
The C-10 carbonyl in 202, having served its purpose was then removed by Wolff-Kishner reduction. After N-methylation of 203, the amide carbonyl was removed by hydride reduction. Hydration of the isolated π-bond in 204 proceeded completely regio and stereoselectively to give 205 in "yields up to 28%". This fortunate selectivity is understandable in terms of a stereoelectronic preference for anti periplanar diaxial addition to the C=C bond with addition of the nucleophile preferentially syn to the protonated amino substituent. The original intention had been to demethylate both ether groups of 205 and to attempt a selective remethylation of the less sterically congested 3-hydroxyl. The action of pyridinium hydrochloride, however, not only cleaved both phenolic ether groups but also dehydrated the secondary alcohol. Fortunately some demethylation of the C-4 methyl ether had been observed during the 202 to 203 conversion. This discovery was exploited by developing conditions that afforded 206 in 54% yield upon heating with KOH in ethylene glycol. Presumably releif of steric congestion fosters demethylation of the 4-methoxy group by an SN2 displacement of phenolate by hydroxide. Completion of the morphine skeleton by generating a furan ring required considerable adjustment of functionality and stereochemistry in 206. A carbonyl at position 6 could be exploited both to allow activation of the 5-position toward intramolecular nucleophilic attack by the C-4 hydroxyl and to allow epimerization at the 14 position. Thus, oxidation of the C-6 hydroxyl in 206 by a variation of the Oppenauer reaction gave ketone 207. To provide the conjugation with the C-6 carbonyl needed to allow epimerization at C-14, a C=C bond was introduced between carbons 7 and 8. Thus, bromination followed by Mattox-Kendall de-hydrobromination (see section 5.4) provided the epimerized tosylhydrazone 198.

After hydrolysis to an enone 210 and reduction to a ketone 211, activation at the 5-position was achieved by bromination with two equivalents of bromine. It is not clear why 207 could not be converted directly into 212 without the intermediacy of 210 and 211. Subsequent monodehydrobromination of an intermediate α,α'-dibromo ketone with DNPH delivered 212. This underwent cyclization in pyridine to yield benzofuran 213 after hydrolysis of the hydrazone. During the 207 to 208 conversion an adventitious bromo group was introduced into the A ring. This was conveniently removed during the reduction of the C-6 carbonyl with lithium aluminum hydride to give codeine (190), the monomethyl ether of morphine (171). Hydride delivery to 213 occurred stereoselectively from the more sterically accessible convex face. Demethylation of 190 was achieved by nucleophilic displacement by chloride of phenol from the protonated ether.
A Friedel-Crafts B Ring Annelation Strategy
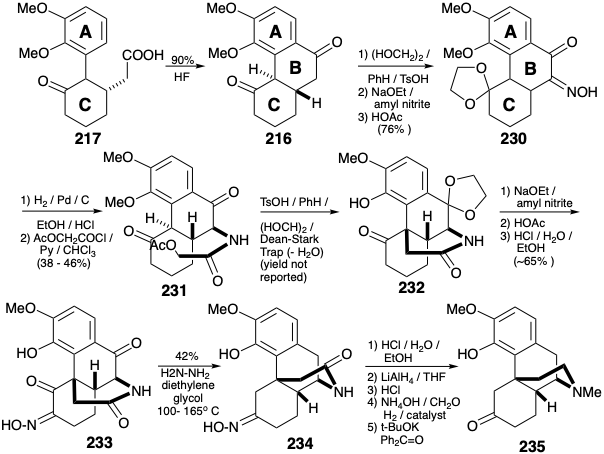
Another strategy for synthesis of morphine (171) is channeled by the decision to generate the B ring by electrophilic substitution of an electron-rich A ring nucleophile.11 This tactic requires temporary carbonyl functionality at the incipient 10 position that would have to be removed in the final steps, e. g. by reduction of a precursor 214. Target- related oxygen functionality at position 5 can be exploited to facilitate introduction of the remaining C-ring functionality and unsaturation and to facilitate generation of the nitrogen heterocycle by alkylation of a carbon nucleophile at position 13. This requires activation of the incipient carbon 15, i.e. α to the amide carbonyl in 214, with a nucleofuge. Appendage of an amino nucleophile to position 11 in a precursor 216 for 215 cannot be achieved by a polar process since neither keto group in 216 can provide electrophilic activation at the 11 position. On the other hand, a nitrogen electrophile could be added to an intermediate that is nucleophilic at position 11 because of activation by the neighboring ketone carbonyl, e.g. by nitrosation of ketone 216. The ABC-ring carbocyclic skeleton of morphine can be assembled by intramolecular Friedel-Crafts aromatic substitution of an electron rich AC-ring precursor 217. Polar analysis of 217 recommends an a,b-unsaturated enone electrrophile 218 that would provide 217 by addition of a carboxy activated nucleophile.
A precursor 219 of 218 might be generated by electrophilic aromatic substution of 220 by a carbonyl activated electrophile, 1,2-cyclohexanedione. However, steric congestion would favor the alternative regioisomeric product 222. The use of an alternative nucleophile, the regioselectively ortho lithiated aromatic diether 221 as nucleophile, avoids this ambiguity. Alternative routes to 218 are recommended by the possibility of employing cyclohexanone as a more readily available C ring starting material. Thus, the arylcyclohexene 224 could be functionalized by a dihydroxylation-oxidation sequence to give 218 through 219 and 223. The possibility of generating 218 directly from 224 by allylic oxidation suffers from the ambiguity of an alternative regiochemical course leading to 225.
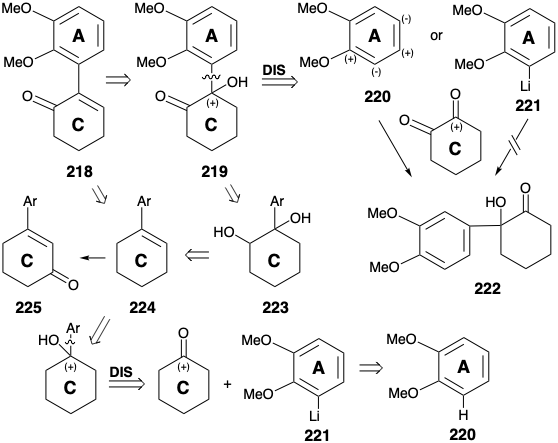
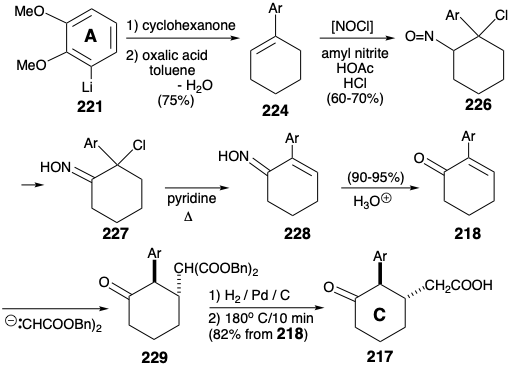
An arylcyclohexene 224 is readily available by ortho lithiation of veratrole (220) with butyllithium and reaction of the resulting aryllithium 221 with cyclohexanone followed by acid-catalyzed dehydration. Allylic bromination (with NBS) or chlorination (with t-butylhypochlorite) followed by hydrolysis and oxidation did deliver the requisite enone 218. But this intermediate was more readily available (overall yields of 40 - 50%) by addition of nitrosyl chloride (from amyl nitrite, acetic acid, and 30% HCl), dehydrochlorination of the intermediate nitrosochloride 226 as the oxime tautomer 227 to the unsaturated oxime 228 and hydrolysis.
The two carbons required to complete the B-ring were appended by Michael addition of dibenzyl malonate carbanion to the enone 218, hydrogenolysis of the resulting 229, and decarboxylation. Friedel-Crafts cyclization of 217 provided the B-ring in 216. Differentiation of the carbonyls in 216 could be accomplished by selective ketalization of the more electrophilic carbonyl. An amino substituent was introduced by nitrosation of an enolate. Reduction of the oxime 230 under acidic conditions was accompanied by deketalization. N-acylation delivered α-acetoxyacetamide 231. Intramolecular alkylation and selective ketalization (now of the less sterically congested carbonyl) occurred upon treatment of 231 with acid. Transposition of the C-ring carbonyl was initiated by nitrosation of the enolate of 232. Deketalization followed by Wolff-Kishner reduction of the intermediate diketo oxime 233 delivered oxime 234 removing two carbonyl groups but not the oxime-masked carbonyl. Hydrolysis of the oxime, reductive removal of the amide carbonyl, reductive methylation of the resulting amine, and oxidation of an intermediate secondary alcohol delivered the ketone 235. Conversion of an analogous intermediate 208 to morphine (171) was described above.
A Conjugate Addition-Alkylation Strategy for B Ring Annulation
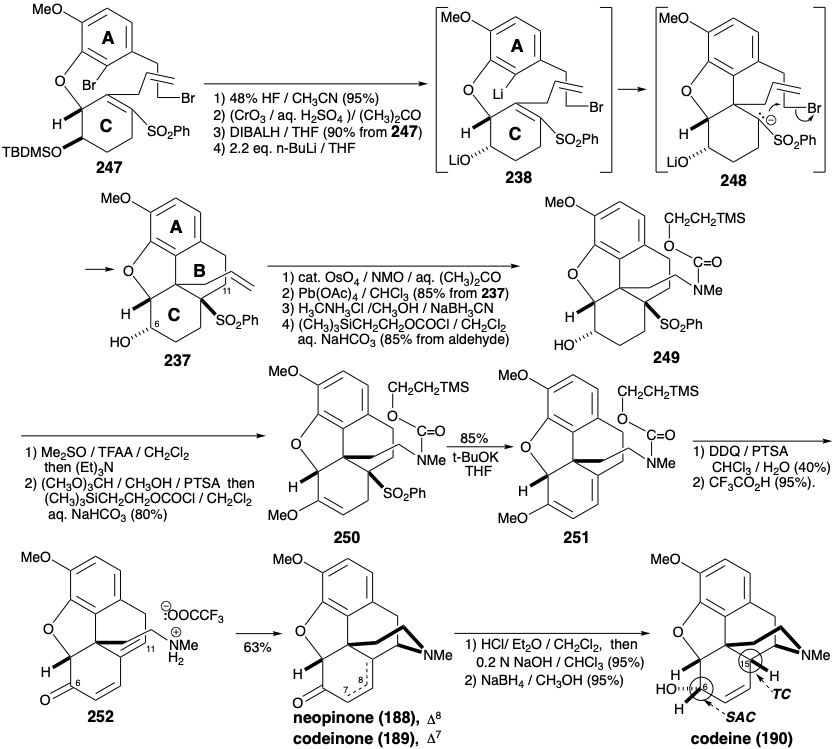
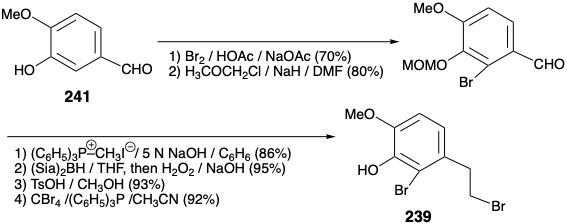
Both of the foregoing syntheses of morphine involve: (1) extensive functional group manipulation after construction of the ABC and piperidine rings, (2) generation of the furan ring last, and (3) a dependence on carbonyl groups to activate or control reactivity. A completely different strategy was employed to achieve a more convergent synthesis of morphine.12 This strategy involves: (1) generation of the piperidine ring last, (2) minimal functional group manipulation after completion of skeletal construction, and (3) exploitation of a sulfonyl group to provide polar activation. As for the biosynthesis and previous syntheses of morphine, the aromaticity of the A-ring recommends an aromatic starting material for this ring. A consonant circuit between C-ring oxygen and B-ring nitrogen substituents in morphine (171) suggests a construction of the piperidine ring, that exploits the polar reactivity provided by target-related functionality in an α,β,γ,δ-unsaturated ketone precursor 236. A polar double disconnection of the B-ring, between a pair of common atoms and between a common and a noncommon atom, is made possible by a strategically placed phenylsulfonyl activating group in a precursor 238 of 237. Polar disconnection of 238 suggests A and C-ring precursors 239 and 240 that should be available from isovanillin (241) and the symmetrical 2-allylcyclohexane-1,3-dione (242) by functional group additions and interconversions.
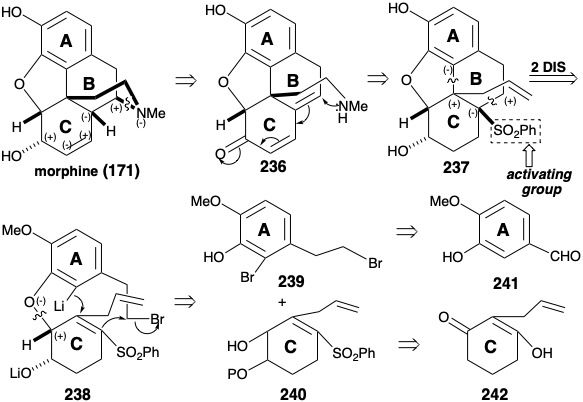
The key A-ring intermediate 239 is available on a large scale from isovanillin (241) in 40% overall yield as outlined below. Substitution of the enolic hydroxyl in 2-allylcyclohexane-1,3-dione (242) by a phenylsulfonyl group is accomplished through the vinylogous acyl chloride 243 to provide 244 by addition of phenylsulfinate and elimination chloride, respectively, in 74% yield overall. Oxidative functionalization of 244 was accomplished by a Rubottom reaction, i.e. treatment of the corresponding enol silyl ether with m-chloroperbenzoic acid. Neither the electron deficient α,β-unsaturated sulfone nor the terminal C=C bond are oxidized in competition with the more electron rich silyl enol ether. Steric approach control in a hydride reduction of 245 delivers the allylic alcohol 246 stereoselectively.
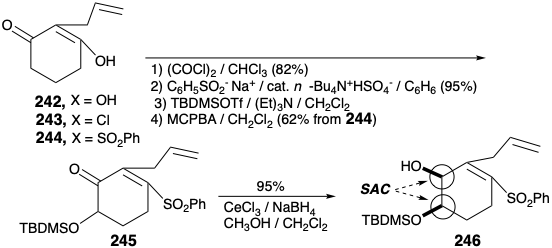
O-alkylation of 246 with 239 provides the key intermediate 247, that undergoes a remarkable cyc- lization upon halogen-metal exchange. Intramolecular Michael addition of the intermediate aryllithium 238 leads via sulfone-stabilized carbanion 248 to 237. Construction of the piperdine ring requires conversion of the allyl group in 237 into an ethylamino sidechain and conjugation of C-11 with the oxygen functionality at position 6 in 237. Oxidation and enol etherification delivers 250 from 249. Elimination of phenylsulfinate to give 251, and hydrolysis to deliver 252 sets the stage for the completion of the morphine ring system by generation of the piperidine ring. Thus, neutralization of the ammonium salt 252 generates an amino group that undergoes spontaneous intramolecular 1,6-Michael addition to the dienone to deliver a mixture of neopinone (188) and codeinone (189) in 63% yield. Conversion of this mixture via codeine (190) to morphine (171) was accomplished as described previously by Rapoport. The required configuration of the hydroxyl at position 6 is established during a steric approach controlled delivery of hydride to the carbonyl carbon in 189. The correct configuration at the 15-position in 190 arises from equilibration through the common enol derivative of ketones 188 and 189.