6.2: Cephalotaxine
- Page ID
- 285466

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The biosynthesis of cephalotaxine (115) involves a convergent strategy that assembles an intricate multicyclic skeleton from two aromatic amino acid precursors, phenylalanine (6) and tyrosine (7). As in the biosynthesis of colchicine (8), one aromatic ring is incorporated intact while the other is extensively modified. Thus in the biosynthesis of colchicine (8) the seven-membered C-ring is elaborated by a one- carbon expansion of a tyrosine-derived aromatic ring. In contrast, the biosynthetic strategy for cephalotaxine (115) exploits a one-carbon ring contraction to produce a five-membered C-ring from a phenylalanine-derived six-membered ring. The logic of the strategy is based on: (1) the ready availability of highly oxygenated cyclohexyl derivatives such as 119 by oxidative metabolism of aromatic precursors and (2) the possibility of extruding a carbon atom as carbon dioxide from an α-diketone by a benzylic acid rearrangement to an α-hydroxy acid. This suggests the α-hydroxy acid 116 as precursor to 115.
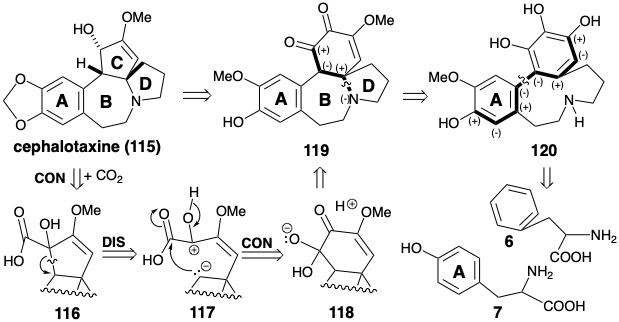
Retrosynthetically, dislocation of a benzylic acid rearrangement product 116 to a precursor 119 corresponds to polar disconnection of the migrating carbon as nucleophile resulting in oxidation of the migration terminus. Subsequent connection of the nucleophilic migrating carbon in 117 results in reduction of the migration origin in the precursor 118. Polar analysis of 119 suggests polar disconnection of nitrogen as nucleophile from an electrophilic carbon β to a carbonyl group. Polar analysis of the precursor 120 suggests that the aromatic rings of two precursors 6 and 7 might be joined by an oxidative coupling.
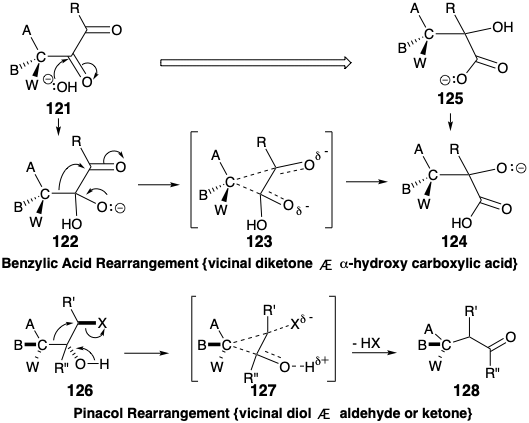
The connection-disconnection sequence of the benzylic acid rearrangement, generalized in the 121 to 125 conversion, is mechanistically analogous to the pinacol rearrangement discussed in chapter 4 (see section 3.4). The rearrangement of 122 into 124, involved in the benzylic acid rearrangement, is isoelectronic with the 126 to 128 conversion of the pinacol rearrangement, i. e. the same electronic movements in identical arrays of atoms, bonds, and nonbonding electrons are involved. Nucleophilic addition of hydroxide to an α-diketone 121 initiates the benzylic acid rearrangement, that proceeds through a temporarily-bridged transition state 123, and ultimately produces an α-hydroxy acid 125. The pinacol rearrangement proceeds through a temporarily-bridged transition state 127. In both the benzylic acid and pinacol rearrangements, the migrating group acts as an internal nucleofuge-nucleophile that adds to an electrophilic migration terminus. In both rearrangements the functionality level of the migration origin increases while the functionality level of the migration terminus decreases.
The biosynthesis of cephalotaxine (115) is believed to involve oxidative coupling of two electron rich aromatic rings in a phenethylisoquinoline6 intermediate 128 delivering a tetracyclic δ-amino-α,β- unsaturated ketone 129. The polar formation and subsequent polar cleavage of a temporary six-membered nitrogen heterocycle in 128, facilitates the oxidative coupling by making it an entropically more favorable intramolecular cyclohexannelation rather than a cyclodecannelation that must generate 130 directly. It is reasonable to postulate the presence of a methoxyl group at position 7 in 128 since this could account for the regioselective oxidative coupling at position 8a which is para to the hydroxyl group presumed to be present at position 6. This regioselectivity contrasts with that observed in the oxidative coupling of autumnaline (28) at position 4a (see section 6.1). Thus, the O-methyl groups in 28 and 128 serve as regiocontrol elements in the oxidative couplings of these phenethylisoquino-lines. Reduction and regioselective methylation of 130 set the stage for regioselective electrophilic activation by oxidation of the ortho hydroquinone 131 to an ortho quinone 132.

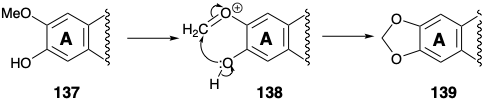
Nucleophilic Michael addition of the secondary amine then delivers 133 whose keto tautomer 134 is an α-diketone. Benzylic acid rearrangement initiated by conversion to 135 delivers α-hydroxy acid 136 in which the carboxyl carbon is derived from a meta carbon of the phenylalanine (6) starting material. Loss of this carbon as carbon dioxide then generates cephalotaxine (115) after conversion of the ortho methoxy-phenol array into a methylenedioxy group. This conversion, i.e. 137 to 139, is common in Nature and presumably involves oxidative generation of an electrophile 138.
B Ring Annelation by Electrophilic Aromatic Substitution
As in the biosynthesis of cephalotaxine (115), the stability of aromatic derivatives recommends the utilization of an aromatic precursor for ring A. The Weinreb strategy for construction of the cephalotaxine skeleton7 recognizes the potential utility of the amino group and dissonant C-ring functionality for activating polar reactions that could append the C-ring onto an ABD-ring precursor. Synthetic equivalents 142 and 143 correspond to the polar synthons 140 and 141. A carbonyl group in 143 is added to facilitate generation of a precursor 145, the amide of prolinol (146) and the arylacetic acid 147. The enamine in 143 could be produced by dehydration of a β-hydroxy amine precursor 144 that, in turn, should be available directly by polar union of an aromatic nucleophile and aldehyde electrophile in 145.
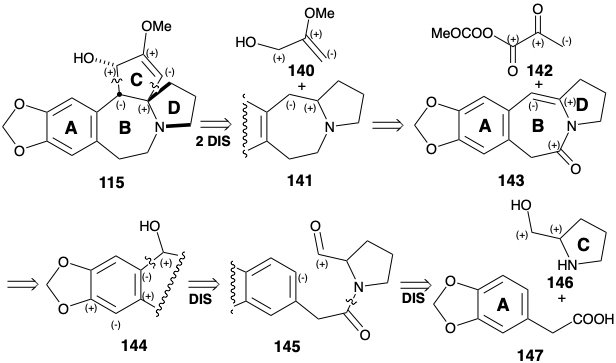
The enamine 143 was constructed by annelation of ring B between an aromatic ring A precursor 147 and a preformed ring D precursor 146. Masking of the hydroxyl group in 146 is unnecessary since acylation occurs at the more nucleophilic nitrogen to give amide 148 rather than at the less nucleophilic oxygen to produce an ester. The final bond of ring B was formed by electrophilic aromatic substitution which occurred exclusively at the less congested aryl position in 145. Having served its purpose, the amide carbonyl was reductively removed from 143 to deliver 149. The polar activation afforded by the acyl group in 142 is first exploited to unite 142 and 149 to give 150. Then the polar activation afforded by both carbonyl groups is exploited to complete the annelation of ring C. An intramolecular Michael addition of an enolate anion to the electrophilic β-carbon atom of an α,β-unsaturated carbonyl system leads to 153. The required cis-ring fusion of ring C is undoubtedly the most stable. The methyl carbonate anion leaving group in 142 is especially noteworthy. Decarboxylation of this anion generates methoxide in situ that then deprotonates an intermediate iminium ion 150 to produce the Michael acceptor 151 under exceptionally mild conditions. Also noteworthy is the use of magnesium methoxide as base to generate the enolate 152 in the Weinreb synthesis of cephalotaxine. Magnesium can assist the cyclization by chelation that enforces a favorable cisoid conformation. Final adjustment of functionality involved enol etherification and hydride reduction. Delivery of hydride occurs from the less sterically congested convex face of 154 producing 115 with the correct relative configuration at the third asymmetric center in ring C. A regioisomeric enol ether was obtained together with 154. This isomer could be recycled by acid catalyzed equilibration.
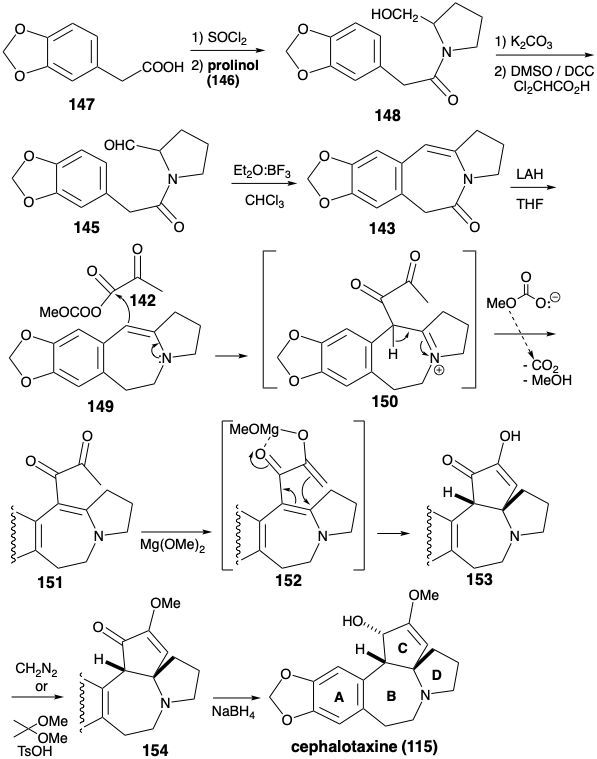
B Ring Annelation by Nucleophilic Aromatic Substitution
Whereas annelation of ring B in the Weinreb synthesis of cephalotaxine (115) was achieved by electrophilic aromatic alkylation, Semmelhack's synthesis creates the same connection by nucleophilic aromatic alkylation.8 Semmelhack's strategy exploits a C-ring carbonyl to provide the requisite nucleophilicity in a final intermediate 155. As in the biosynthesis and the Weinreb strategy, an aromatic precursor is exploited for ring A. N-alkylation of a CD-ring amine fragment 157 with the A-ring fragment 156 provides 155. The same A-ring starting material 147 is used for both total syntheses. Ring C in 157 contains two oxygen functionalities that provide electrophilic activation at their respective carbon atoms. Thus, these functional groups cannot be exploited directly to create the bond between those carbon atoms by a polar reaction. In the Weinreb synthesis of cephalotaxine (115), ring C was constructed by polar reactions by using a starting material 142 that incorporates the dissonant circuit between the two oxygen functionalities in the C-ring. The Semmelhack strategy recognizes that this dissonant circuit in 157 can be formed by a nonpolar reaction, reductive coupling of the two electrophilic carbonyl carbons in a precursor 158. Although 158 might be available directly by polar addition of two carbomethoxymethyl nucleophiles to an electrophilic D-ring precursor 160, Semmelhack opted for the alternative strategy of adding two allyl nucleophiles to 160 followed by oxidative revelation of the latent carboxyl groups in an intermediate 159.
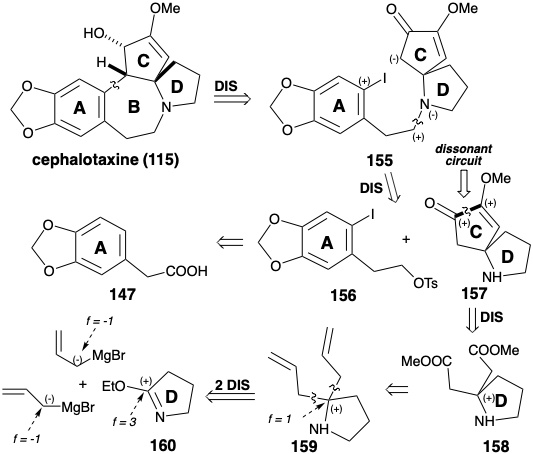
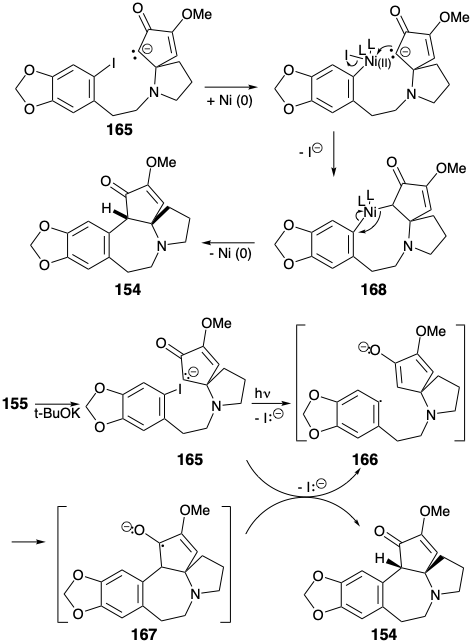
The CD-ring intermediate 157 was prepared from pyrrolidinone. The reaction of an imino ester 160 with an allyl nucleophile gave 159. Masking of the amino group as an acid labile amide 161 was required prior to oxidative cleavage of the C-C π-bonds in 159 which ultimately provided the diester 158. Intramolecular acyloin coupling of 158 in the presence of chlorotrimethyl silane (the Rühlmann modification) produced 162, that was oxidized directly to 163 by a one-pot addition of bromine and elimination of TMSBr. Methylation of this symmetrical dione delivered 157. Alkylation of this amine with the nitrosylate 164 provided 155. Cyclization of 155, vide infra, followed by reduction of the intermediate 154 delivered cephalotaxine (115). This synthetic strategy leads directly to the correct enol ether 154 without formation of the regioisomeric enol ether that is a byproduct in the Weinreb synthesis.
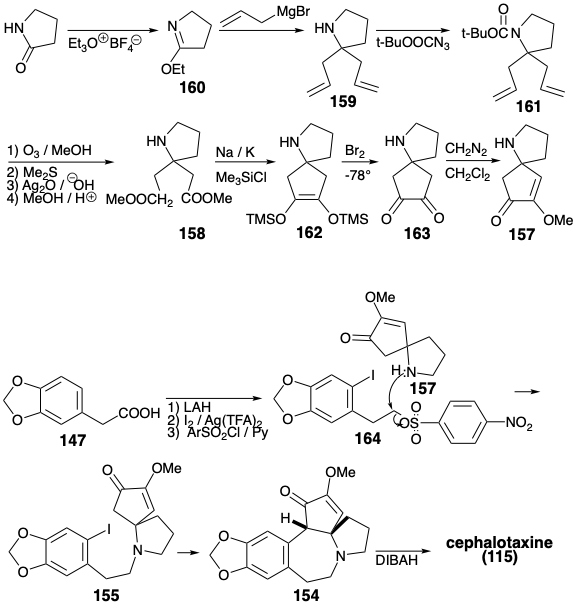
The key cyclization of 155 to 154 was achieved by a variety of reactions all involving nucleophilic aromatic substitution. Of course, direct nucleophilic attack on the electron rich A ring aryl iodide does not occur when an enolate nucleophile is generated from 155. However, net nucleophilic substitution could be accomplished by photolysis of the enolate 165 or by treatment of 165 with Na/K or a nickel(0) catalyst. Best yields of the cyclization product 154 (94%) were obtained by a photostimulated SRN1 reaction presumably involving the chain carrying anion radicals 166 and 167. The SRN1 reaction could also be achieved (45%) by reaction of the enolate 165 with Na/K. A nickel(0)-catalyzed reaction of 165 provided 154 in 30% yield presumably by oxidative addition of the aryl halide to Ni(O) and nucleophilic substitution of iodide by a carbanion producing a σ-aryl-nickel(II) intermediate 168, that undergoes reductive elimination of 154 to regenerate the Ni(0) catalyst.