
3.3: Syntheses of Prostaglandins from Acyclic Precursors
- Page ID
- 285444

As in the biosynthesis of prostaglandins, several total syntheses feature generation of the cyclopentane ring by cyclization of acyclic precursors. We will first compare the biosynthesis with three total syntheses involving cyclopentane ring formation by generation of the prostaglandin 8,12-bond. Since the 8,12-bond lies on dissonant circuits between the functionality at positions 9 and 11 or at positions 9 and 15, a polar connection cannot be achieved which depends on polar activation by either pair of functional groups. Therefore, each of these syntheses exploits the polar activation provided by only one target-related functional group to promote polar creation of the 8,12 bond. In Miyano's strategy1 (see below), polar disconnection of the lower side chain of PGE1 at the C=C bond suggests subtarget 19. Neither the aldehyde group nor functionality at C-11 in 19 can provide the requisite electrophilicity at C-12 for polar bond formation exploiting nucleophilicity at C-8 activated by the C-9 carbonyl. Therefore, a precursor of 19 must incorporate additional functionality to provide electrophilicity at the incipient C-12. This might be provided by a carbonyl group as in 20. However this strategy is flawed since the electrophilic aldehyde in 20 will compete with the carbonyl at the incipient C-12 in 20 for reaction with a C-8 nucleophile. This will produce an undesired benzoquinone byproduct derived from the intermediate 21 by dehydration.
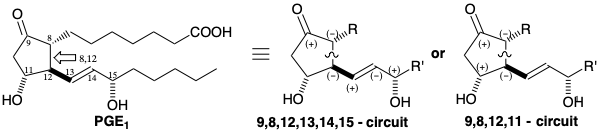
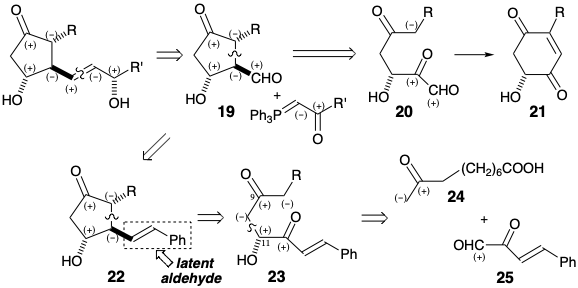
To prevent undesired competition by an electrophilic aldehyde, this group is not introduced until it is needed. Instead, it is concealed in a pre-cursor 22 of 19 as a C=C bond from which it can be generated by oxidative cleavage. The C=C bond does not possess the high electrophilicity of an aldehyde. Yet an aldehyde group can be readily generated from the C=C bond. Polar disconnection of 22 suggests electrophilic functionality at the incipient prostaglandin C-12 in a precursor 23. Further disconnection of 23 to 24 and 25 exploits the polar activation provided by consonant functionality at the incipient C-9 and C-11 ignoring the carbonyl at C-12 in 23.
We shall refer to an unreactive direct precursor group with a different functionality level than the target as a latent functional group (see below for some examples).2 Thus, (1) an alkene (f = 0), as in 22 above, or a vicinal diol (f = 1) can serve as a latent equivalent of an aldehyde or ketone (f = 2). Although the alcohol is electrophilic at the incipient carbonyl carbon, its electrophilicity is considerably less than that of an aldehyde or ketone. (2) An arene (f = 0) is a latent carboxyl group (f = 3). (3) An enol ether (f = 1) provides a latent ester (f = 3) since oxidative cleavage of the latent precursor can be achieved to provide the desired functional group. (4) A ketone (f = 2) is a latent ester (f = 3) since Baeyer-Villiger oxidation of the former will deliver the latter; and (5) a terminal alkyne (f = 0) is a latent acetylide anion (f = -1) or terminal vinyl carbanion (f = -1) equivalent since alkynyl hydrogen is readily abstracted by strong bases and the alkyne products from coupling of acetylides with electrophiles can be selectively reduced to the corresponding alkene. Note that generation of a functional group from its latent equivalent, by definition, involves oxidation [O] or reduction [R], i.e., increase or decrease of functionality level. Also note that generation of a carbanion by proton abstraction from carbon or hydride addition to an α,β- unsaturated carbonyl compound corresponds to a reduction. In other words, proton abstraction from a “carbon acid”, i.e., an acidic C-H bond, corresponds to reduction in functionality level of the carbon from which a proton is abstracted and oxidation of the hydrogen that is abstracted as a proton. Also note that addition of a proton to a C=C bond results in reduction of the proton and and oxidation of the carbon that becomes a carbocation, and addition of \(\ce{Cl2}\) to a C=C bond corresponds to oxidation of both carbons with concomitant oxidation of both chlorines, a process that is called “dioxidative addition”.
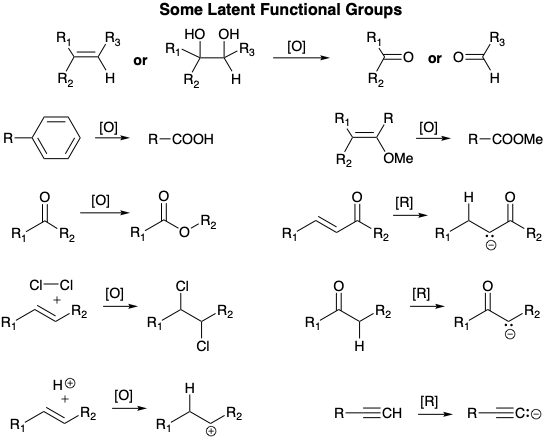
A closely related concept is the masked functional group, which is an unreactive precursor group with the same functionality level as the target. For example, a ketal is a masked ketone (f = 2). Another example is provided by esters (f = 3) that serve as masked carboxyl groups (f = 3). Esterification blocks the proclivity of the acid toward decarboxylation when β to a carbonyl or toward deprotonation by bases. As we shall see in the Kojima-Saki synthesis of prostaglandins (see below), different masking groups for the same functional group, such as a benzyl and a methyl ester for two carboxylic acid groups in a molecule, may allow selective removal (unmasking) to convert one ester to an acid while the other remains masked.
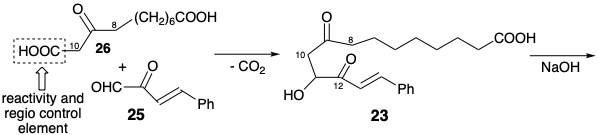
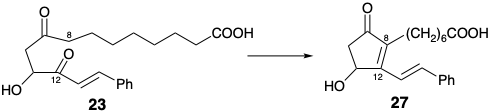
In Miyano's synthesis of PGE1, β-ketoacid 26 rather than methyl ketone 24 (see above) is condensed with ketoaldehyde 25. This decarboxylative condensation regioselectively achieves nucleophilic activation at the incipient 10-position without competition from condensation at the incipient 8-position that also can be activated by the C-9 carbonyl. Thus, the carboxyl group appended to position 10 in 26 serves as an activating group. It is a reactivity control element and, consequently, a regiocontrol element. Intramolecular aldol condensation then generates the 8,12-bond delivering cyclopentenone 27. Selective oxidative cleavage of the more electron rich styryl C=C bond in 27 delivers an unsaturated aldehyde 28. Saturation of the remaining C=C bond affords 29 in which the aldehyde and carboxyheptyl groups adopt the required trans relative configurations owing to a thermodynamic preference and epimerizability at both positions 8 and 12. Condensation of 29 with ylide 30 then delivers PGE1 after selective reduction of the 15-carbonyl. Neither the stereocenter at position 15 nor that at position 11 is generated with high stereocontrol: thermodynamic control (TC). Furthermore, this synthesis produces a racemic mixture of PGE1 and its unnatural enantiomer since any synthesis which begins with nonasymmetric or racemic starting materials and employs racemic reagents will ultimately generate only racemic products.
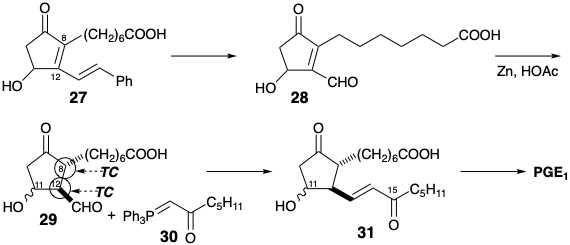
A second strategy3 for prostaglandin synthesis involving cyclopentan-one annulationtion by formation of the 8,12-bond starts with polar disconnection of the lower side chain of PGF2α to suggest a subtarget 32. However, instead of using a C-8 nucleophile and C-12 electrophile as in the Miyano synthesis, the Kojima-Saki synthesis achieves polar 8,12-bond formation by reaction of a C-8 electrophile with a C-12 nucleophile. The C-12 aldehyde in 32 or an ester in a precursor 33 can activate carbanion generation at C-12. To preclude β-elimination of the C-ll oxygen and provide additional stabilization of a C-12 carbanion, the C-11 oxygen is present as a carbonyl group in the precursor 33 of 32. A possible β- elimination of the C-9 oxygen is precluded by concealing this functional group in a latent form as a benzyloxycarbonyl group in 33. Polar disconnection of 33 requires electrophilic reactivity at C-8 which could be provided by conjugation with the benzyloxycarbonyl or an additional carbonyl group at C-8 as in 34. This carbonyl group also can provide nucleophilic activation at C-9 for polar generation of the 9,10 bond. The benzyloxycarbonyl also can aid in formation of a carbanion at C-9 and prevent interference by carbanion generation at C-7. Thus, the benzyloxy-carbonyl also serves as an activating group and a regiocontrol element. The 9,10,11-circuit in 34 is dissonant. Therefore, polar disconnection at a nucleophilic C-9 requires electrophilic activation of the incipient C-10 in a precursor 36. The carbonyl at C-11 in 34 is ignored in the polar disconnection to 36 which is an umpoled synthon from methyl acetoacetate (37).
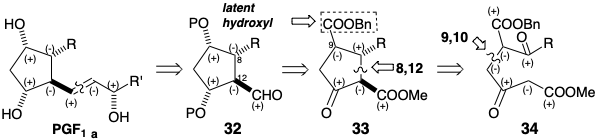
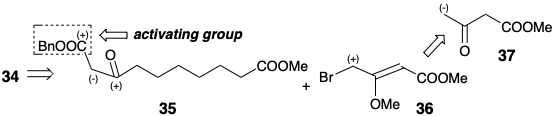
As in the 23 to 27 cyclization of the Miyano synthesis, cyclization of 34 produces a cyclopentanone 38. But now the roles of nucleophile and electrophile are reversed. Thus, in the Miyano synthesis the nucleophilic center is at position 8 and the electrophilic center at position 12 in 23.
In the Kojima-Saki synthesis, the nucleophilic center is at position 12 and the electrophilic center at position 8 in 34. This intermediate is stereoselectively converted to 33 by catalytic hydrogenation which saturates the C=C bond and also selectively converts the benzyl ester into a carboxylic acid without affecting the methyl or ethyl esters. Preferential generation of the α,α,β relative configurations at C-9, 8, and 12 respectively in 33 results from steric approach control followed by epimerization of the β-keto carbomethoxyl group. Conversion of 33 into PGF1α is straightforward.
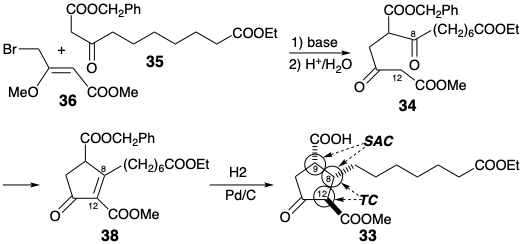
Generation of the C-10 hydroxyl from the latent precursor, a C-10 benzyloxycarbonyl, is achieved by Baeyer-Villiger oxidation of an intermediate methyl ketone 39. Selective reduction of the carboxyl in 39 at C-12 is achieved by masking the electrophilic reactivity of the C-1 carboxyl as a potassium salt. To allow selective manipulation of the resulting alcohol, the hydroxyls at positions 9 and 11 in 40 are masked as tetrahydropyranyl (THP) ethers prior to reduction of the carbomethoxy group.
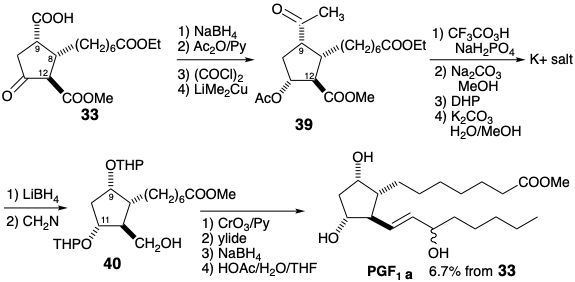
For a synthesis of PGE1 from 39, generation of the C-9 oxygen functionality from its latent precursor is delayed until after the C-11 acetoxy group in 39 is converted into a THP ether to allow differentiation from the C-9 acetoxy generated in a Baeyer-Villager oxidation of 43. The dithioketal protecting group in 41 can be selectively removed from 42 after introduction of the THP ether. Addition of the lower sidechain to 43 after oxidation to aldehyde 44 parallels the sequence described for the Miyano synthesis.
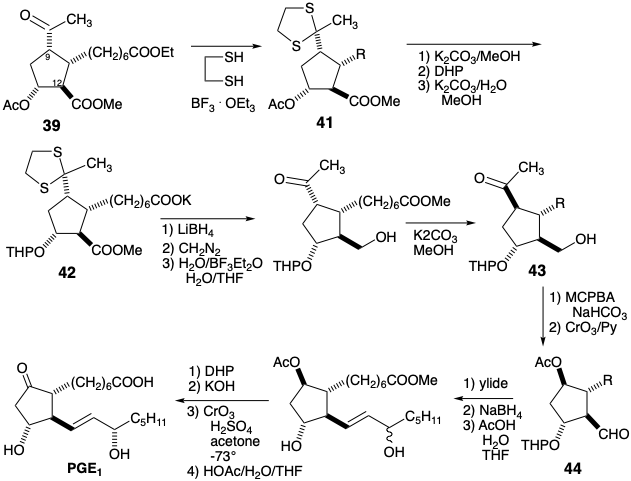
The β-hydroxycarbonyl array in PGE2 is sensitive toward dehydration. If the C-9 carbonyl is to be exploited to activate nucleophilic reactivity at C-8 in a precursor, this sensitivity must be addressed. In the Miyano synthesis, dehydration of the cyclization product 27 is disfavored by the instability of the antiaromatic cyclopentadienone that would result. In a third strategy for prostaglandin synthesis4 that also mimics the biosynthetic cyclopentane annulation involving 8,12-bond creation, the problem is circumvented by introducing the C-11 hydroxyl at the end of the synthesis. This suggests a key intermediate 45 in which the C-11 functionality is removed and an activating group is added at C-8 to aid in carbanion generation at this position. Polar disconnection of the upper side chain from the subtarget 45 suggests electrophilic functionality at C-7 as in the precursor 50. Polar disconnection of 46 at the 8,12- bond reveals the need for electrophilicity at C-12. However, rather than placing appropriate functionality at C-12 as in intermediate 23 of the Miyano synthesis (see above), a nucleofuge at C-14 in 47 provides the requisite activation by conjugation with C-12. An epoxide 47a provides the appropriate electrophilicity at C-12 and oxygen functionality at C-15. A chlorohydrin 47b is an alternative synthetic equivalent that is interconvertible with 47a. Polar analysis of 47b suggests disconnection to the electrophilic precursor 49 and 48 whose terminal acetylene serves as a latent vinyl nucleophile. The utility of terminal acetylenes as latent terminal vinyl nucleophiles also suggests a strategy for a C-C connective route to 50 via 51 from 52, cyanide, and a 1,3-propane dielectrophile. Although 47a and 47b are alternative synthetic equivalents, conversion of 47b to 47a during the synthesis would provide a more reactive electrophile.
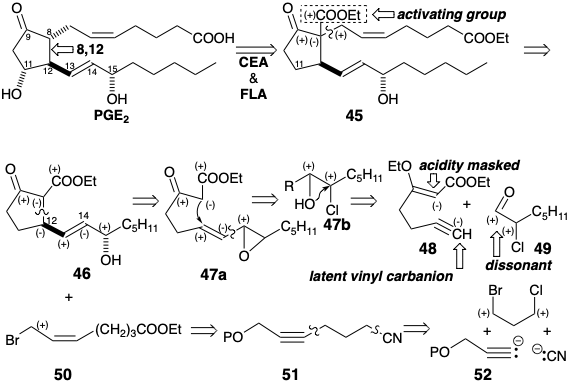
A synthesis of 48 might have been achieved by propargylation of ethylacetoacetate dianion. However, the potential for abstraction of the acetylenic hydrogen, by the strongly basic dianion, recommended an alternative approach.
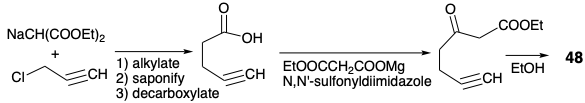
Thus, a malonic ester synthesis provided 4-pentynoic acid that was further elaborated to a β-ketoester by Claisen condensation.4 Enoletherification then masks the ketone in 48 and allows selective deprotonation at the terminal acetylene. Condensation of the resulting acetylide nucleophile with α-chloroheptanal (49) produces 53 stereoselectively and then cis vinyl trans epoxide 47a after partial hydrogenation and base promoted heterocyclization.
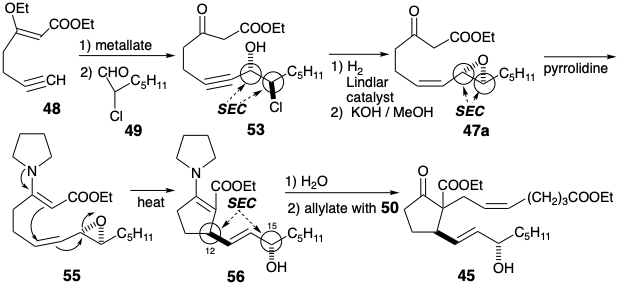
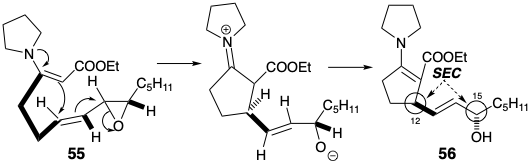
Cyclization of β-keto ester 47, generating the prostaglandin 8,12-bond, might have been accomplished by intramolecular alkylation of an intermediate enolate. However, an alternative nucleophile, enamine 55 was employed. Hydrolysis of an intermediate enamine derivative 56 followed by allylation of the resulting β-ketoester delivered 45. Especially noteworthy is the stereoselectivity of this synthesis of 45 with the correct relative stereochemistry at C-12 and C-15. This is the consequence of three consecutive stereoelectronically controlled (SEC) reactions.
First, addition of an acetylide nucleophile derived from 48 to the α-chloroaldehyde 49 occurs stereoselectively at the least hindered face of the carbonyl group in a conformation of 49 that achieves maximum separation of the C=O and C-Cl dipoles by an anti periplanar arrangement. Subsequent cyclization of the β-chloroalkoxide 54 with Walden inversion produces a trans disubstituted epoxide stereospecifically.
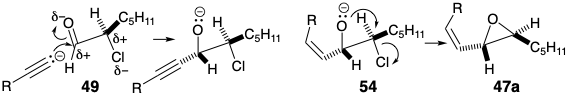
A stereoelectronically preferred mode of cyclization was expected to translate the stereochemical relationship between the chiral centers in the cis vinyl trans epoxide 55 into the requisite stereochemical relationship between the chiral centers at positions 12 and 15 in 56.4 Thus, anti SN2' displacement of alkoxide by the enamine nucleophile in 55 was expected to generate 56 after proton transfer in a presumed iminium alkoxide intermediate.
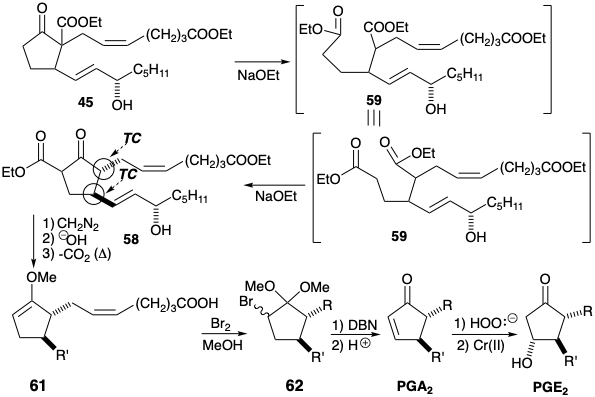
Introduction of a hydroxyl substituent at the 11-position in 45 is complicated by an absence of activating functionality adjacent to C-11. Reactivity can be provided by introducing unsaturation between carbons 10 and 11 taking advantage of the C-9 carbonyl to activate C-10 for introducing a leaving group. PGE2 could then be obtained by epoxidation of the α,β-unsaturated ketone PGA2 followed by reductive cleavage of the C-O bond adjacent to the carbonyl in 57. This suggests PGA2 as a synthetic precursor of PGE2.
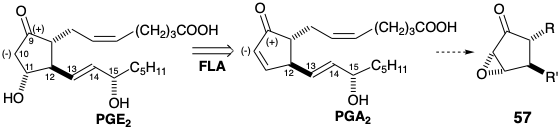
While 45 (see above) might be converted to PGA2 by decarbethoxylation of the β-ketoester array and subsequent oxidative introduction of a leaving group at C-10, a more elaborate strategy was adopted. The process provides an example of a construction of the prostaglandin skeleton by generating the 9,10-bond of the cyclopentane ring as the last skeletal connection. Thus, a 10-carboethoxy precursor 58 would be well suited for the regioselective (α vs α’) nucleophilic activation (proton abstraction) of the 10-position required for the conversion of a 10,11-dihydro-PGA2 derivative into PGA2. Also, 58 is suitably functionalized for polar generation by reaction of a C-10 nucleophile with a C-9 electrophilic center in a precursor 59. Polar reconnection of 59 suggests the key intermediate 45 as a precursor of 58 via 59.
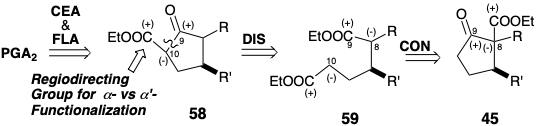
PGA2 is a naturally occuring dehydration product from PGE2, and the former has been prepared from the latter by an oxidation reduction sequence. Stereoselectivity during the epoxidation was achieved by employing a sterically demanding blocking group appended to the 15-hydroxyl in 60 to direct epoxidation to the α face of the cyclopentenone ring.5
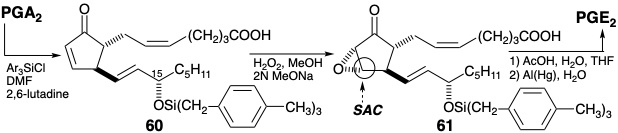
The conversion of 45 into 58 was achieved by an ethoxide-catalyzed rearrangement presumably involving retro Dieckmann cleavage to 59 followed by Dieckman cyclization to 58. The carboethoxy transfer might also occur by decarboethoxylation and recarboethoxylation. In any event, the rearrangement is driven to 58 by formation of the corresponding enolate. The rearrangement allows regiocontrol in the formation of an enol ether 61, the subsequent introduction of bromine by 1,2-dioxidative addition to give 62, and ultimately in the introduction of unsaturation between carbons 10 and 11.
The previous three examples of prostaglandin synthesis illustrated polar synthesis of the 2,3-bond of a cyclopentanone ring corresponding to the 8,12-bond of the prostaglandin skeleton. In each case, this requried additional functionalization of a precursor because the 9,8,12,13,14,15 or 9,8,12,11-circuits are dissonant. Since the 9,10,11 circuit is consonant, either the 9,10 or 10,11- bond can be formed by a polar reaction that depends only on target-related functionality. As we shall see in section 3.7, seco prostaglandins (molecules lacking one C-C bond of the prostaglandin skeleton) are natural products for which the name levuglandin (LG) was coined to signify that they are derivatives levulinaldehyde with prostaglandin side chains. In theory, 13,14-dihydro-PGE1 (63) could be generated directly from dihydro-LGE1 by polar reaction of a C-10 nucleophilic enolate with the electrophilic carbonyl carbon of the aldehyde group at C-11, i.e an aldol condensation. However, although 11,13-dihydro-PGE1 is undoubtedly an intermediate in this cyclization, it is not stable and dehydrates under the basic aldol condensation reaction conditions to afford olefin 64, that must then be refunctionalized to deliver the target β-hydroxyketone 63.6
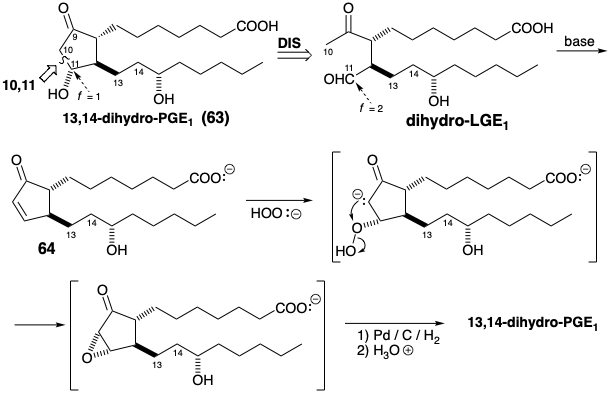
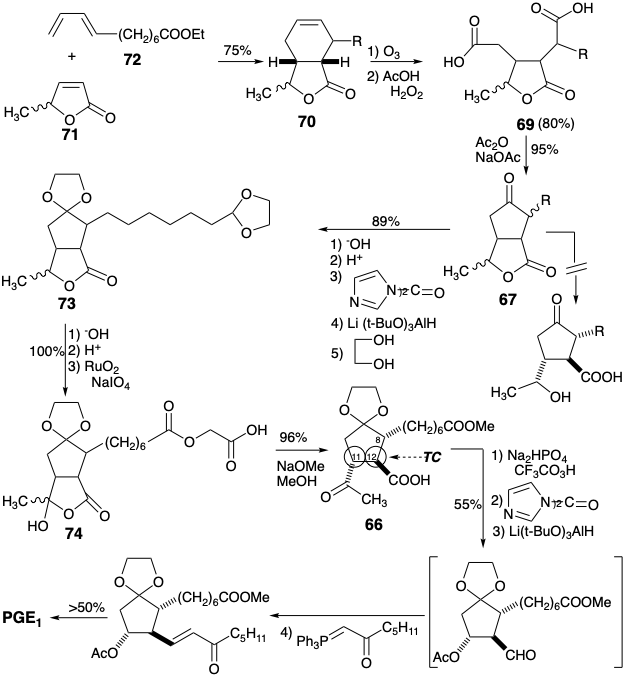
A Merck synthesis7 of PGE1 achieves cyclopentanone annulation by creating the 9,10-bond in a polar process. However, to avoid generation of a sensitive β-hydroxy ketone intermediate, only one target related functional group is exploited. Polar disconnection of the lower side chain suggests a precursor 65 (see below). The C-11 hydroxyl is concealed in latent form until the end of the synthesis to avoid β-elimination. Thus, subtarget 65 is dislocated to a penultimate precursor 66 in which the C-11 hydroxyl is replaced by a methyl ketone and the C-9 carbonyl is masked to allow selective Baeyer-Villiger oxidation of the methyl ketone to deliver 65. The reactive methyl ketone and carboxylic acid functionality in 66 can be internally masked as a lactone in a precursor 67. Although the ring juncture in 67 is necessarily cis, the substituents at both positions 11 and 12 in 66 and its stereoisomers are epimerizable allowing generation of the required all-trans relative configuration of the substituents in 66 that is favored thermodynamically.

To facilitate polar synthesis of the cyclopentanone ring in 67, a carboxyl group can be appended to the incipient C-10 in a precursor 69 to provide nucleophilic activation (CEA). This activating carboxyl will be removed readily from the cyclization product 68 by a polar cleavage. The two reactive carboxylic acid groups in 69 can be derived from a latent precursor 70 by oxidative cleavage. Finally, the cyclohexene moiety in 70 suggests a double disconnection to 71 and 72 that would provide the cyclohexene ring by a Diels-Alder cycloaddition. A preference for the required orientation of the cycloaddition is predicted owing to orbital overlap effects which favor an ortho relationship between the substituent on the diene and the electron withdrawing substituent on the dieneophile.
During the synthesis, it was discovered that opening of the lactone in 67, to allow oxidation of the secondary hydroxyl and deliver ketone 66, could not be achieved. To circumvent this flaw in the original plan, 67 was converted to an acetal 73 that could then be converted into a hemiacylal 74 by \(\ce{RuO4}\) oxidation. Transesterification replaced the carboxymethyl ester in 74 with a methyl ester in 66 that could then be converted to PGE1 as planned.
In the above synthesis, a cycloaddition was applied to generate a cyclohexene by a C-C double connective process. Cycloadditions generally provide a valuable nonpolar route to various cyclic products. Functionality is unnecessary for cycloadditions. Usually the targets contain unsaturation, but cyclobutanes, that can be generated by cycloaddition, do not even have unsaturation. A retro cycloaddition (RC) dislocation is feasible if a cyclic shift of two σ-bonds and n-2 π-bonds cleaves the ring into two poly and/or monoene precursors. Targets that are candidates for synthesis by cycloaddition of two poly and/or monoenes can be recognized by the presence of at least n-2 π-bonds in a 2n-atom closed circuit. If the precursors are bridged, the cycloaddition will be intramolecular. Cycloadditions may occur readily with thermal activation if n is an odd number but may require photochemical activation or transition metal catalysis if n is an even number. These rules are a consequence of the requirement for positive overlap between the lowest unoccupied molecular orbital (MO) of one reactant with the highest occupied molecular orbital (HOMO) of the other reactant. Sometimes this is only possible for the HOMO of the photoexcited π* state of one reactant and usually the interaction is suprafacial (on the same face of the π molecular orbitals. However, note that for 2π + 2π cycloadditions, a thermal reaction is favorable if one of the π-MOs interacts antarafacially (on opposite faces) and undergoes twisting of the two ends of the MO in opposite directions during the cycloaddition. For steric reasons, this is usually not feasible. However, it occurs readily for ketenes (see section 3.4).
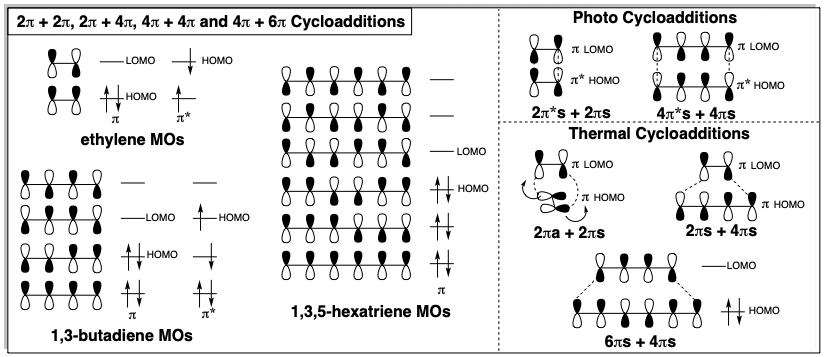
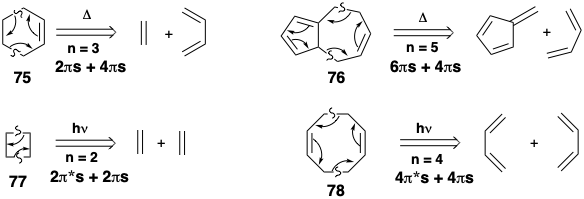
Some examples of retro cycloaddition dislocations are presented below. The precursors of 75 and 76 may cycloadd with thermal activation while the precursors of 77 and 78 may require photoactivation, transition metal catalysis or involve antarafacial reaction of one π-bond, e.g., of a ketene.
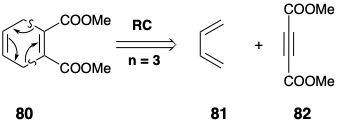
The 2n atom circuit may contain more than n-2 π-bonds. For example, dislocation of 80 to 81 and 82 corresponds to a cycloaddition that will readily occur thermally. Shift of two σ-bonds and n-2 = 1 π-bond cleaves the ring into two polyenes and n = 3 is an odd number.
Since the 11,12,13,14,15-circuit in PGE1 is consonant, in theory annulation of the cyclopentanone ring by forming the 11,12-bond can be achieved by exploiting the polar activation provided by the functional groups at positions 11 and 15. For example, conjugation with the C-15 carbonyl in 84 should facilitate generation of a carbanion at C-12 that could couple with the aldehyde carbonyl in 84 to generate 83 directly. However, PGE1 contains a sensitive β-hydroxycyclopentanone array that readily dehydrates to give PGA1. Therefore, early strategies for the synthesis of PGE1 were dominated by efforts to mask the reactive β-hydroxy ketone array.
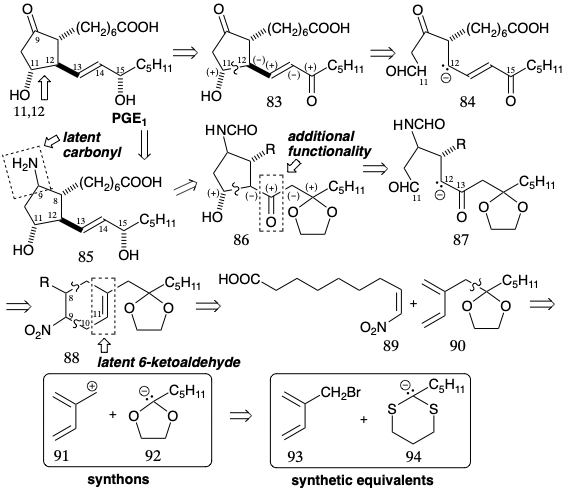
In the first total synthesis of PGE1, achieved by E. J. Corey8, the C-9 ketone carbonyl was carried along in latent form as a relatively unreactive formamide grouping which was transformed into a carbonyl group only at the end of the synthesis, vide infra. For the ultimate skeletal connection between carbons 11 and 12 in a precursor 87 of 85 in Corey's strategy for PGE1, nucleophilic reactivity at C-12 is provided by a carbonyl group added to C-13. This carbonyl would be removed after polar cyclization to 86. Furthermore, reduction followed by β-elimination fostered by a carbonyl group at C-15 would generate the required unsaturation between carbons 13 and 14. The two reactive carbonyl groups in 87 could be derived from a latent precursor, the C=C bond in a cyclohexene 88. A double disconnection of the latter intermediate suggests diene 90 and dienophile 89 as precursors that would provide 88 by a Diels-Alder cycloaddition. The use of a nitro group in 88 and 89 as precursor for the formamido group in 87 is dictated by the favorable reactivity of electron deficient dienophiles toward electron rich dienes such as 90. For the synthesis of diene 90, the polar union of an electrophilic isoprenoid synthon 91 with an acetal carbanion synthon 92 was chosen since isoprenyl bromide 93 is readily available. A dithiane-derived carbanion 94 would serve as the synthetic equivalent of the umpoled ketal carbanion synthon 92. The allylic bromide 93 was prepared by free radical allylic bromination of 95, a cycloadduct obtained from isoprene and sulfur dioxide. Free radical abstraction of allylic hydrogen is favored by delocalization of the resulting allylic radical. Benzylic hydrogen abstraction and bromination with N- bromosuccinimide (NBS) is simillarly favored. Both 95 and the derived allylic bromide are crystalline solids from which the correspond-ing dienes can be generated by cycloelimination of \(\ce{SO2}\). Allylation of 94 with 93 delivered a diene 96 that is a synthetic equivalent of 90 (see above).
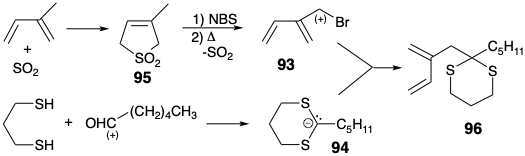
Rather than first convert thioketal 96 into ketal 90, the former was reacted with dienophile 97 to produce cyclohexene 98. Then the dithioketal was replaced with an ethylene ketal protecting group. Revelation of the latent carbonyl groups by oxidative cleavage of the alkene 88 provided the ε-keto aldehyde 87. Base catalyzed aldol cyclization of 87 then delivered 86 stereoselectively. Thus, because the C-12 substituent is epimerizable in 86, the required thermodynamically favored trans relationship between the two bulky side chains was produced. Because no control was exerted during generation of the C-11 stereocenter, some of the wrong epimer was also formed.
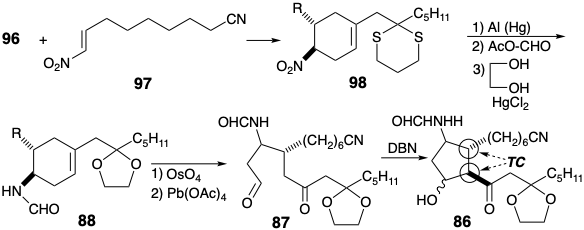
Although the prostanoid carbon skeleton was generated in the aldol cyclization of 87 to give 86, completion of the synthesis required extensive adjustment of functionality and unsaturation level. The sensitive β-hydroxy ketone array was immediately acetylated and then the carbonyl group was reduced to avoid dehydration. After deprotection of the ketone and dehydration of the intermediate 99, the allylic carbonyl in an intermediate enone was reduced with \(\ce{Zn(BH4)2}\), a mild hydride reducing agent, to produce an allylic alcohol 100. Hydrolysis of the nitrile and acetate was followed by protection of the hydroxyl groups at positions 11 and 15 as tetrahydropyranyl ethers. Subsequent hydrolysis of the formamide required more vigorous conditions to deliver a THP protected derivative of 85.
Production of PGE1 by generation of the sensitive β-hydroxy ketone array under mild conditions was then accomplished in the key step of the synthetic plan. The strategy to use an amino group as a latent carbonyl in the immediate precursor 85 of PGE1 depended upon a precedented sequence of reactions. Thus, selective oxidation to an imine was accomplished by N-bromination with NBS followed by base promoted elimination. Finally, hydrolysis of both the imine and the tetrahydropyranyl (THP) ether protecting groups under mildly acidic conditions delivered PGE1.
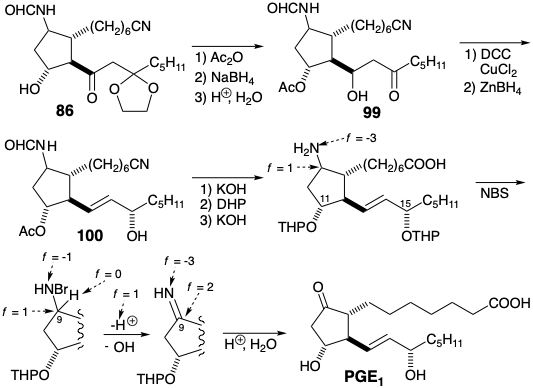
The key oxidation process is based entirely on polar reactions. Thus, N-bromination oxidizes the amino nitrogen from f = -3 to f = -1. Elimination of \(\ce{HBr}\) then reduces the nitrogen to f = -3 while it oxidizes carbon and hydrogen.