3.4: Syntheses of Prostaglandins from Polycyclic Precursors
- Page ID
- 285445

In the preceding syntheses of prostaglandins stereocontrol was achieved in several ways. For example, stereoselective generation of 33 from 38 depended on steric approach control (SAC) during catalytic hydrogenation to favor a cis relationship between substituents at positions 9 and 8. Subsequent thermodynamic control (TC) favored a trans relationship between the substituents at positions 9 and 12 by epimerization of a thermodynamically less stable cis intermediate to the more stable trans isomer. Stereoelectronic control (SEC) was adduced to account for the stereoselective generation of the correct relationship between the stereocenters at positions 12 and 15 in 56 during the cyclization of 55.
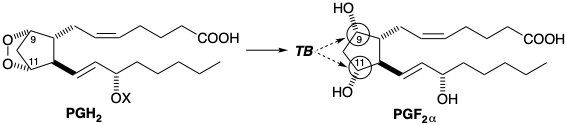
In this section, another technique for achieving stereocontrol will be considered. Thus, proximity of functional groups may be assured by the tactic of tying them together in a temporary ring which is ultimately cleaved. The biosynthesis of PGF2α involves such a temporary bridge (TB) that enforces a cis relationship between the oxygen atoms at the 9 and 11 positions. These oxygens are tied together with an O-O bond in the intermediate prostaglandin endoperoxide PGH2.
Often, temporary bridges contain functional groups in a latent form. We previously saw that cycloolefins may be oxidatively cleaved to yield dicarbonyl compounds. Creation of the PGF2α skeleton by generation of both C=C bonds from carbonyl precursors suggests a 1,5-dialdehyde subtarget 101. Since the aldehyde substituent at the C-12 stereocenter should be epimerizable, the less thermodynamically favored all cis aldehyde 102 can also serve as subtarget. The carbonyl groups in 102 can be concealed in latent form in the temporary unsaturated bridge of 103. By employing a second temporary unsaturated bridge, a highly stereocontrolled synthesis of PGF2α can be achieved. Thus, the cis-1,3-diol array found in PGF2α and in the proposed intermediate 103 can be obtained -- by Baeyer-Villiger oxidation of the derived methyl ketone - from the cis-diacid 104 that can be produced by oxidative cleavage of endo-dicyclopentadiene (DCPD). In this strategy for prostaglandin synthesis9, generation of the required cis relationship between the stereocenters at positions 8 and 9 ultimately depends upon a stereoelectronic preference for generation of the endo rather than exo isomer of DCPD during \(2 π + 4 π\) cycloadditive dimerization of 1,3-cyclopentadiene. This is favored by secondary orbital overlap between the "nonparticipating" C=C bond and the cycloadding diene.
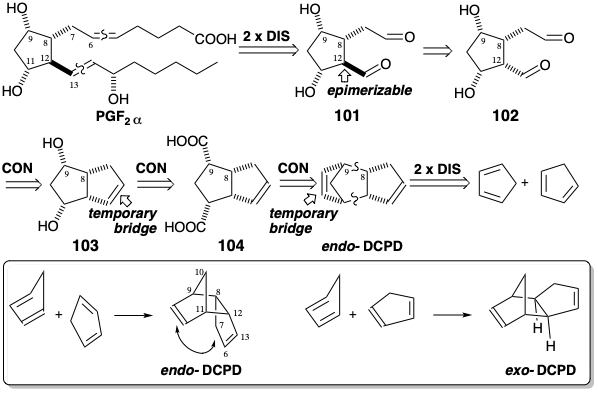
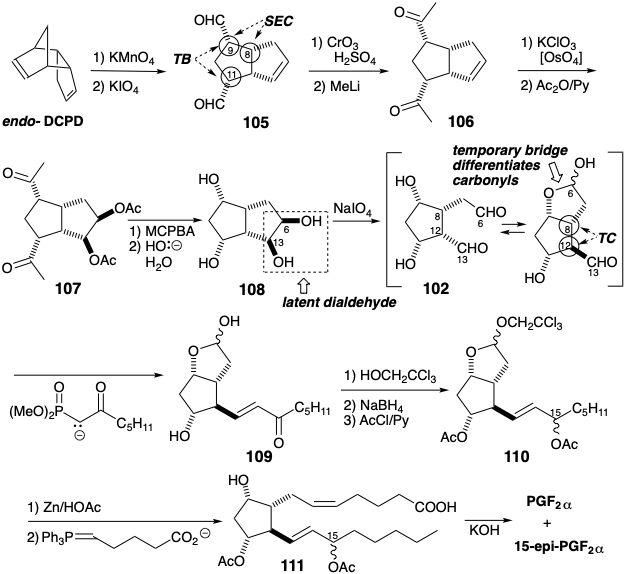
This synthesis exploits selective cleavage of one temporary bridge, the more strained C=C bond, to produce a dialdehyde 105. After conversion to diketone 106, the remaining C=C bond is partially oxidized to deliver 107 after acetylation. Baeyer-Villiger oxidation then produces a tetraacetate. Saponification followed by oxidative cleavage provides dialdehyde 102 from its latent precursor, the vicinal diol array in 108. Epimerization of 102 at position 12 generates 101 that forms a hemiacetal in which one aldehyde carbonyl is adequately masked to allow chemoselective olefination of the remaining carbonyl to provide 109. To prevent reduction of the aldehyde carbonyl, 109 is converted to a mixed acetal before a nonstereoselective reduction of the ketone carbonyl. Reductive cleavage of the β-trichloroethyl acetal in 110 then allows olefination of the remaining aldehyde group to provide 111 and ultimately racemic PGF2α together with the racemic 15-epimer.
Corey's second strategy for total synthesis of prostaglandins10 exploits two temporary bridges to assure the proper stereochemical relationships between the stereocenters at positions 9, 8, and 11. The cis relationship between the substituents at positions 8 and 9 in 101 is assured by a temporary ring in the lactone precursor 112 that is generated by stereoselective functionalization of an olefin 113. Another temporary bridge, invloving the C-8 substituent, is used in the lactone 114 to assure a cis relationship with the hydroxyl at position 11. Since ketones are latent esters the cyclic ketone 115 can serve as a precursor of 114. The required regiospecificity in the Bayer-Villiger oxidation of ketone 115 can be expected since this reaction involves 1,2-migration to an electron deficient terminus. The group that most readily supports a partial positive charge migrates preferentially. Thus, in 115 the secondary alkyl group that is also allylic migrates in preference to the primary alkyl group. A trans relationship between the substituent at position 12 and the remaining stereocenters in the cyclopentane ring is ultimately the consequence of steric approach control during the cycloaddition of a 5-substituted 1,3-cyclopentadiene precursor 116 with ketene.
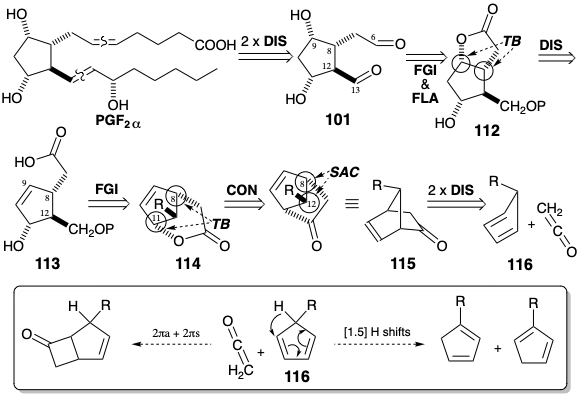
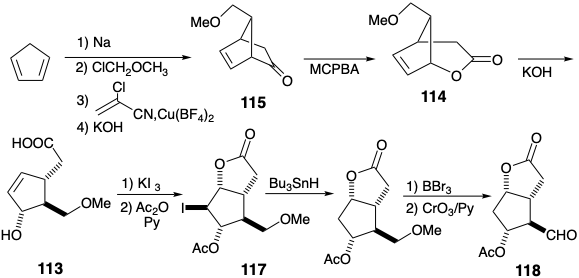
A potential flaw in this strategy arises from the instability of 5-substituted 1,3-cyclopentadienes that readily rearrange at room temperature, by [1.5]sigmatropic hydrogen migrations (see section 4.7), to generate mixtures of 1 and 2-substituted isomers. This isomerization was circumvented by using \(\ce{Cu(BF4)2}\) to catalyze the Diels-Alder reaction of 5-methoxymethyl-1,3-cyclopentadiene with α-chloroacrylonitrile at low temperature (to avoid rearrangement of the 5-substituted 1,3-cyclopentadiene). This chloronitrile -- a latent ketene -- undergoes 2π +4π cycloaddition with cyclopentadiene whereas ketene prefers to undergo 2πa + 2πs cycloaddition (see section 3.3). Another potential flaw, epoxidation of the C=C bond in 115 during Baeyer-Villiger oxidation, is apparently prevented by steric shielding of the C=C bond. Stereoselective (one configuration is generated preferentially at a new stereocenter) introduction of the C-9 hydroxyl results from the stereocontrolling influence of a temporary bridge. Thus, a nucleophile that is appended to C-8 in 113 is introduced intramolecularly (internal nucleophile) to an electrophilic center created at C-9 by the addition of I\({}^{\oplus}\) to the C-C π-bond to give 117. Deprotection and oxidation generate the aldehyde 118.
Appendage of the lower side chain to aldehyde 118 is achieved by olefination with a β-keto phosphonate. Reduction of an intermediate ketone followed by transesterification gives a lactone diol in which the hydroxyl at position 9 is differentiated from the remaining hydroxyls. The latter are then masked as tetrahydropyranyl (THP) ethers in 119. Partial reduction of the lactone and Wittig olefination of an intermediate aldehyde delivers a key intermediate 120 that can be converted into E or F prostaglandins of the "1" or "2" series by approptiate manipulation of protecting groups to allow selective adjustments of functionality and unsaturation levels. Thus, the hydroxyl at position 9 in 120 or the derived 122 can be selectively oxidized to afford PGE2 or PGE1 respectively while deprotection of 120 or 122 delivers PGF2α or PGF1α respectively.
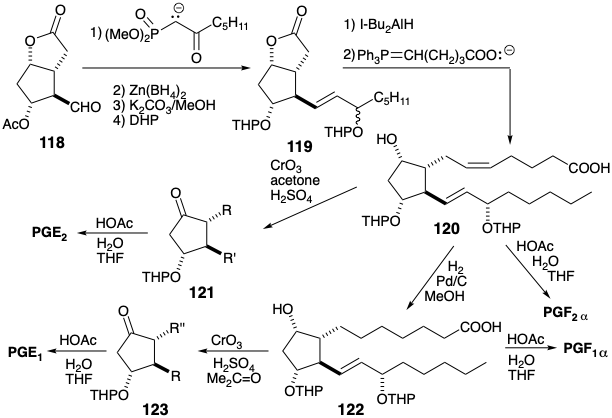
A closely related strategy for synthesis of prostaglandins11 exploits exactly the same temporary bridges to enforce a cis relationship between the substituents at positions 9, 8, and 11 on the cyclopentane nucleus. However, a different order for generating the same skeletal connections obviates the necessity of using protecting groups. Thus, the upper side chain is added to a precursor 124. However the lower side chain is already present in a bicyclo[2.2.1]heptene intermediate 125 prior to Baeyer-Villiger cleavage of the temporary bridge. Also the necessity for a difficult low-temperature Diels-Alder cycloaddition is avoided by using a fulvene 127 instead of a 5-substituted cyclopentadiene to react with a ketene equivalent 128. The presence of an aldehyde enol acetate in 126 and 127 also avoids the requirement for a subsequent adjustment of functionality level after hydrolytic removal of the masking group.
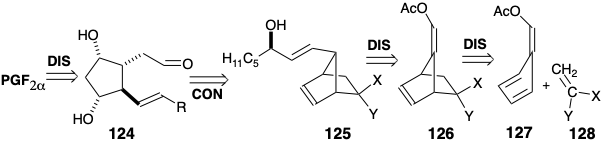
The enol acetate in 129 is readily hydrolyzed selectively in the presence of the α-chloronitrile. The stereoselective generation of the requisite configuration at the incipient 12 position is undoubtedly the consequence of thermodynamic control. Thus, the aldehyde adopts the least sterically congested configuration. After olefination of this aldehyde with a β-ketophosphonate carbanion, the ketone carbonyl is reduced by a Meerwein-Pondorf-Verly reaction followed by hydrolysis of the α-chloronitrile delivering 130 in good overall yield. Since this ketone incorporates appreciable ring strain, an unusual Bayer- Villiger-like cleavage with hydroperoxide anion is possible. Peracids react with 130 to give epoxides, but the hydroperoxy anion reacts exclusively with the carbonyl group. The diol 131 can be masked to eventually allow selective oxidation of the 9-hydroxyl delivering PGE's while further elaboration to PGF2α closely follows the synthesis from 120 except that THP protecting groups are unnecessary. Because no steps involving introduction or removal of protecting groups are required, this synthetic strategy is remarkably efficient.

The strategies presented thus far all succeeded. Unfortunately, failed attempts are often not published. Sometimes they are described in doctoral theses. It would be a mistake to assume that synthetic planning for the total synthesis of complex molecules is so dependable, even by the undisputed superstars of organic synthesis. Therefore, to maintain a realistic perspective, we will consider some flawed strategies from time to time.
R. B. Woodward’s Flawed Strategy
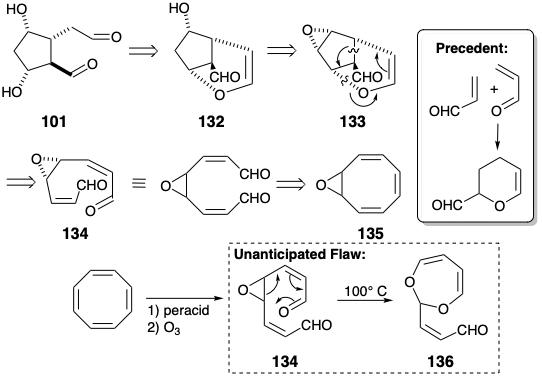
The dialdehyde 101, that incorporates all the stereochemical information required for the cyclopentane ring of prostaglandins, might be derived from an enol ether 132 in which two functional groups are internally masked in a temporary bridge. The use of an epoxide 133 as a precursor of the alcohol 132 is recommended by the consequent possibility that 133 can be generated from a symmetrical precursor 134. This strategy requires the discovery of a method to achieve regioselective reductive cleavage of epoxide 133 to generate 132. The presence in 133 of a six-membered ring containing one C=C bond suggests the possibility of a 2π + 4π cycloaddition that would generate 133 from 134. The intramolecular hetero Diels Alder cycloaddition of an aldehyde dieneophile to an α,β-unsaturated aldehyde diene to generate the dihydropyran ring in 133 is precedented by the corresponding intermolecular dimerization of acrolein which, however, favors the wrong orientation. Generation of 134 might be feasible by selective oxidative cleavage of the most electron rich C=C bond in cyclooctatetraene monoxide 135. This strategy, devised by Woodward, is fatally flawed because the intermediate 134 undergoes a novel homo retro Claisen rearrangement producing 136 instead of the desired Diels-Alder cycloaddition.1
Ring Contraction Strategies
The mixed acetal 137 is another temporarily bridged precursor similar to Corey's aldehyde 118 (see above) except that the functionality level in 137 is identical with that in 101. Therefore, the temporary bridge in 137, like that in 132, is an internally masked derivative of two functional groups as opposed to a latent precursor. Woodward12,13 recognized that a potential precursor of 137 is 138 in which C-11 and the aldehyde carbonyl carbon are temporarily bridged by a C-C bond. The intermediates 137 and 138 are isomers with different connectivities and a different distribution of functionality but identical overall functionality level. The rearrangement of 138 to 137 involves oxidation to an aldehyde of a carbon bearing a hydroxyl and concomitant reduction of a carbon bearing an electronegative leaving group. Such a process, a pinacol rearrangement, is driven to completion by the energetically favorable generation of a C=O double bond at the expense of two C-O single bonds. The subtarget 138 can be simplified by dislocation to a less functionally substituted precursor 139 that might provide 138 by 1,2-dioxidative addition. Generation of the C=C bond in 139 from a ketone in 140 is suggested by the goal of disconnecting and functionally reorganizing this subtarget to a symmetrical precursor 141 by polar dislocation involving disconnection of an internal electrophile. The ketone carbonyl in 140 can provide nucleophilic reactivity to facilitate this C—C bond formation. A temporary bridge in 141 between the electrophile and nucleophile assure the necessary cis relationship between the masked hydroxyls in 141 and the newly created C-C bond in 140. Ultimately, 141 might reasonably be available by selective protection of two of the three identical hydroxyl groups in all cis 1,3,5- cyclohexanetriol and oxidation of the remaining hydroxyl.
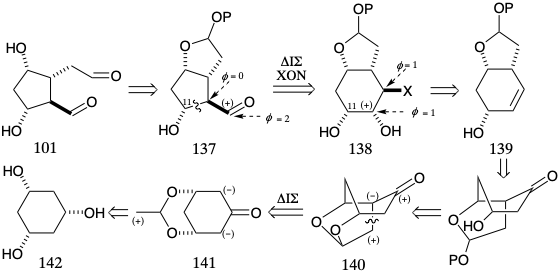
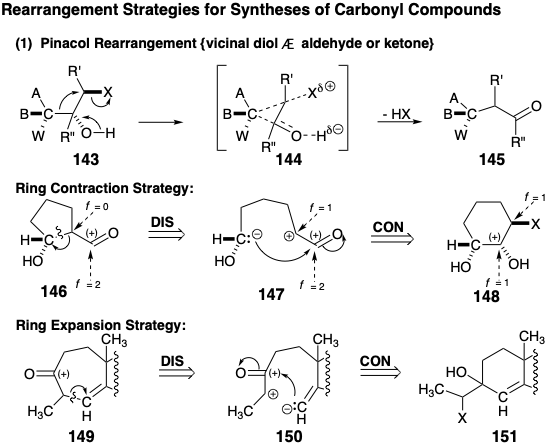
Stereospecific (the stereochemical configuration of the product isomer is determined by that of the reactant isomer) generation of the stereochemical relationships required in 137 is expected during the pinacol rearrangement of 138. Thus, such rearrangements, i.e. 143 to 145, generally proceed with retention of configuration at the migrating carbon owing to a temporarily-bridged transition state 144.
Two Step Reterosynthetic Analysis of Polar Rearrangements
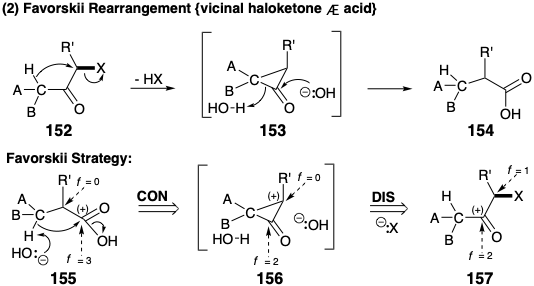
Dislocation of a pinacol rearrangement product 146 to a precursor 148 may be viewed as polar disconnection of the migrating carbon as nucleophile resulting in oxidation of the migration terminus. Subsequent connection of the nucleophilic migrating carbon results in reduction of the migration origin (note that this is an internal nucleophile). In fact, the disconnection and connection steps occur simultaneously in pinacol rearrangements. Synthetically the 148 to 146 rearrangement results in ring contraction. Pinacol rearrangements can also result in ring expansion. This is exemplified retrosynthetically by generation of a precursor 151 with a six-membered ring for a target 149 with a seven-membered ring by disconnection to 150 and subsequent connection to 151. This example, which also suggests that a vinyl carbon may serve as the migrating group, is a step in a strategy for total synthesis of the terpene longifolene that will be considered in chapter 4. Of course, pinacol rearrangements may also occur in acyclic systems. The Favorskii rearrangement of α-haloketones to generate ring-contracted or acyclic carboxylic acids is structrually and functionally related to the pinacol rearrangement. However, the Favorskii rearrangement involves a temporarily-bridged intermediate rather than transition state. Thus, 1,3-elimination from 152 generates a cyclopropanone intermediate 153 from which a ring-contracted product 154 is formed by nucleophile-induced cleavage. Retrosynthetically, Favorskii rearrangements generate a more connected precursor 156 from a carboxylic acid target 155. Disconnection of the precursor 156 then suggests the skeletally and functionally reorganized precursor 157 in which the functionality level of the carboxyl group (f = 3) equals the sum of the functionality levels of a ketone carbonyl (f = 2) and a carbon bearing an electronegative leaving group (f = 1). Thus, as in the pinacol rearrangement, the Favorski rearrangement results in no net change in molecular functionality level, i. e. no net oxidation or reduction. Rather, these processes involve redistribution of functionality by an intramolecular redox process. Thus, rearrangement dislocations are complex because they involve coupled connection and disconnection steps as well as an associated redistribution of functionality.
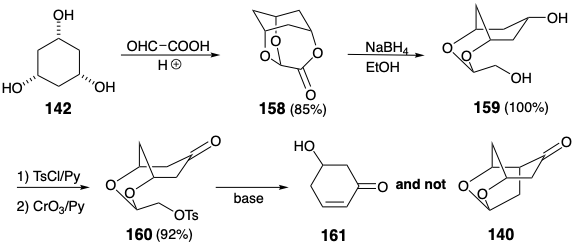
Although differentiation of the three hydroxyls in 142 was achieved by reaction with glyoxalic acid, the second Woodward strategy was also fatally flawed (see section 3.5). Reductive cleavage of 158 afforded diol 159 quantitatively, and the primary hydroxyl in 159 was readily activated selectively by tosylation. However, the ketone 160, obtained by oxidation of the monotosylate, failed to produce 140 upon treatment with a variety of bases. Instead, elimination of a β-alkoxy group to produce 161 occurred to the complete exclusion of intramolecular alkylation.
To obviate the necessity for enolate generation β to the alkoxy groups as in 141, 162 was recognized as a direct precursor to olefin 139. Polar disconnection of 162 suggests addition of a carbon electrophile to a C=C double bond as in 163. The two dislocations, 140 to 141 and 162 to 163 are isoelectronic (mechanisms with identical electron movement patterns). That is, they both involve the movement of two pairs of electrons and the cleavage of a C-C σ-bond. However, whereas cleavage of a C- O bond in a synthetic equivalent of 141 to give 161 is driven by the production of a C=O bond, the similar cleavage of a C-O bond in a synthetic equivalent of 163 would generate a relatively unstable intermediate, an allylic carbocation.
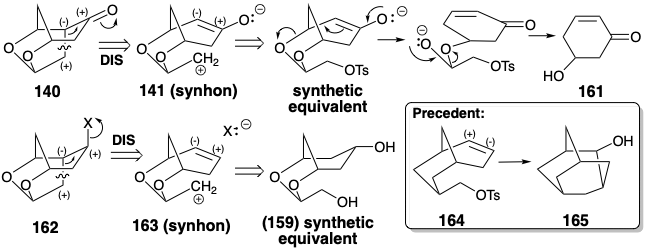
Synthetic equivalents of the unsymmetrical synthon 163 might be obtained from the available symmetrical diol 159 by β-elimination. Unfortunately, a precedent suggested that cyclization of 163 might not occur in the desired fashion. Thus, the carbocyclic analogue 164 produces the isomeric ring system 165 upon solvolysis. However, it could be argued that the allylic oxygen substituents in 163 might disfavor such a mode of cyclization that must generate an electron deficiency β to the alkoxy substituents.
This time the gamble payed off.13 The bismesylate 166 from 159 afforded olefin 167 upon selective elimination of the secondary mesyloxy group. Solvolysis of 167 produced a mixture that contained only 5-8% of the undesired product 168. Of course, the desired cyclization product 169 is racemic. It can be resolved. However, only one enantiomer leads to prostaglandins of natural configuration. Thus, while this synthesis (vide infra) solves "the main sterochemical problem inherent in prostaglandin F2α synthesis -- the alignment of the four contiguous chiral atoms in the cyclopentane moiety", the process is not enantioselective. Half of the intermediate 169 is the wrong enantiomer, that is not readily recyclable. Dehydration of the appropriate cyclization product 169 then provided 170. The requisite stereochemical preference during 1,2-dioxidative addition to 170 was best achieved with the perimidic acid generated in situ from benzonitrile and hydrogen peroxide. This reaction delivered a mixture of the exo epoxide 171 and the endo epoxide 172. The former epoxide could be recycled to 170. Stereoelectronically controlled nucleophilic opening of the epoxy ring in 172 delivered 173 stereospecifically. This isomer is mandatory for the subsequent pinacol-like ring-contracting rearrangement that ensues upon deamination of the bicyclic mixed acetal 174. Thus, migration of the incipient C-11 carbon is favored in 174 by an anti periplanar relationship (two bonds or groups lying in the same plane with a dihedral angle of 180°) with the leaving group. Rearrangement of 174 occurs not only with retention of configuration at the migrating carbon, but also stereospecifically generates the requisite configuration at C-12 in the product 175 owing to Walden inversion during the intramolecular SN2 reaction. Further elaboration of the key intermediate 175 into prostaglandins followed well-precedented reactions.
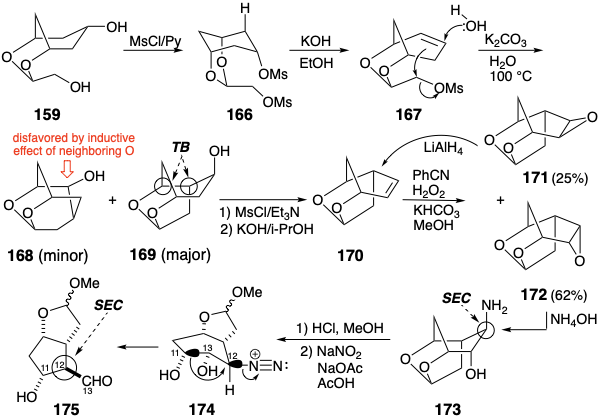
Corey devised an alternative route14 to key intermediates of the type 138. Thus, 138 might be available by cis-1,2-dioxidative addition to alkene precursor 176. Furthermore, a trans relationship between the incipient leaving group X and the newly introduced oxygen atoms might be anticipated on the basis of steric approach control. Steric approach control might also favor the required trans relationship between X and the lactone ring. This would be especially true if allylic oxidation were achieved by an ene mechanism which would involve a sterically demanding cyclic transition state. Furthermore, such a mechanism would assure the requisite regiocontrol, i.e., substitution with allylic rearrangement, during introduction of X. The lactone in 177 could be obtained from a latent precursor, the ketone in 178, available in turn by a 2π + 2π cycloaddition of ketene to a symmetrical precursor, 1,3-cyclohexadiene.
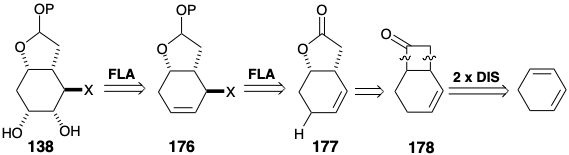
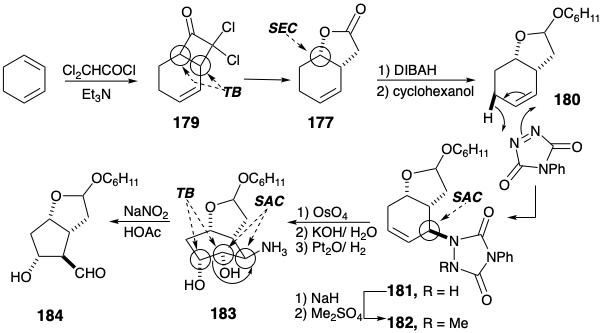
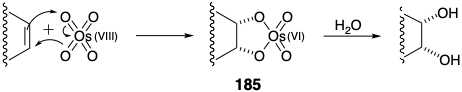
Structurally selective cycloaddition (see section 3.3) of dichloroketone to 1,3-cyclohexadiene delivered 179 from which the unsaturated lactone 177 was obtained by reductive dechlorination followed by Baeyer-Villiger oxidation (see section 3.5). Partial reduction followed by ketalization delivered 180 that stereoselectively afforded 181 upon ene reaction with N-phenyltriazolinedione. Hydroxylation of the derived 182 also proceeded stereoselectively. The latter reaction also involves a temporary ring, an osmate ester 185, that enforces a cis relationship between the newly introduced oxygen atoms.