9: Strategies in Reserpine Synthesis
- Page ID
- 21849

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
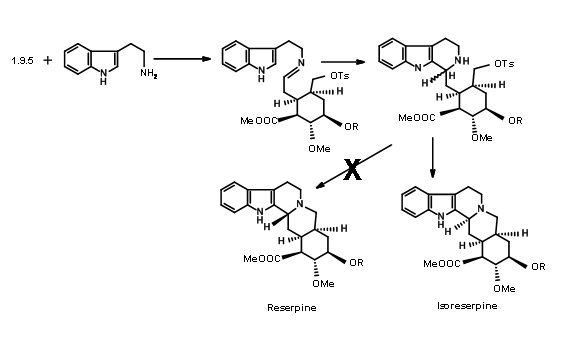
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The structure of Reserpine was solved by 1953. R.B. Woodward’s group reported the first synthesis of Reserpine in 1956 (J. Am. Chem. Soc., 78, 2023, 2657 (1956); Tetrahedron, 2, 1 (1958)). His scholarly analysis clearly displayed aspects of retroanalysis, which was just evolving at that time. This synthesis commands admiration for the way he used conformational analysis and stereoelectronic effects to precisely develop the stereopoints in this exceedingly complex problem for that time. He recognized that the E-ring has a dense array of 5 asymmetric centers in a six membered E ring. His disconnection of reserpine led him to the key intermediate C. We could formalize his retroanalysis as shown in Figure 9.1.
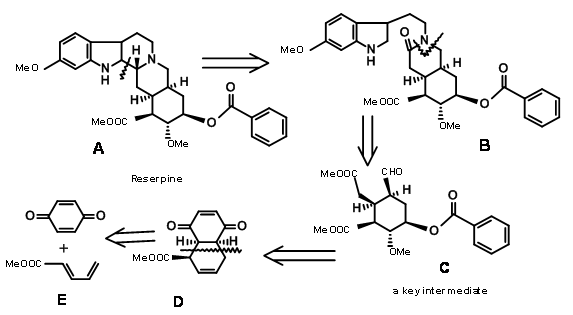
Another brilliant piece of conformational analysis could be seen in the way he converted Isoreserpine to Reserpine by introducing conformational strain in an otherwise comfortable molecule.
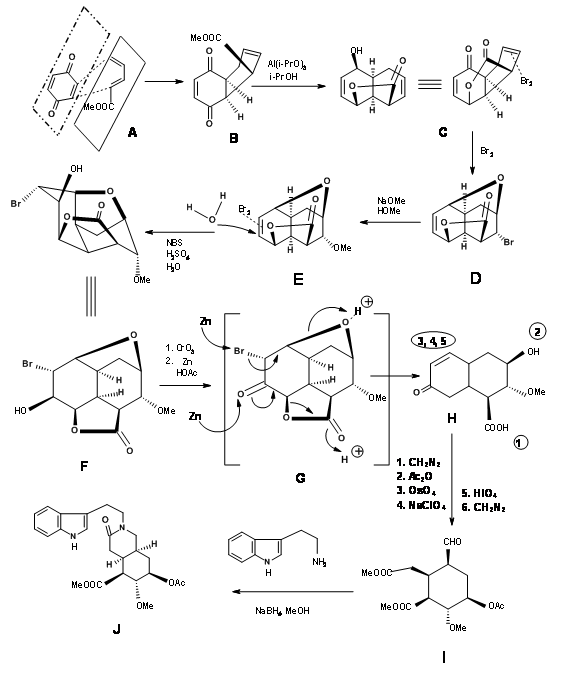
Synthesis of Woodward’s Aldehyde (9.1C): In a cleaver execution by Woodward’s group (Fig 9.2), all the required carbons for the D/E rings and three of the five asymmetric centers were created by one Diels-Alder reaction (9.2B). Note the simple dissymmetry in one component – methyl acrylate – could precisely place three asymmetric centers in a row in a correct fashion. This cycloaddition reaction developed a concave phase and a convex phase1 in the product that guided further modifications on this intermediate. The hydride reagent in the next step delivered the hydride from the less hindered convex phase, placing the –OH group in the concave phase. This facilitated the formation of a five-membered lactone ring (9.2C). The alternate six-membered lactone ring was presumably more strained. Bromination on 9.2C with molecular bromine formed the bromonium ion complex from the convex phase, while the –OH group could enter from the concave phase to form an ether (9.2D). Treatment with sodium methoxide displaced the bromine from the convex phase, possibly via elimination – addition route (9.2E). The next bromonium ion complex again proceeded from the convex phase, with the water molecule entering from the concave phase to enable a trans-diaxial opening of the bromonium complex (9.2F). After one oxidation step, the molecule was now set for a complex double elimination reaction, with the zinc attacking two centers. An attack at the bromide center opened the ether ring while another attack at the carbonyl group opened the lactone ring. This complex Tandem Reaction placed all the five asymmetric centers. Opening the unsaturated ring gave the key intermediate 9.2I.
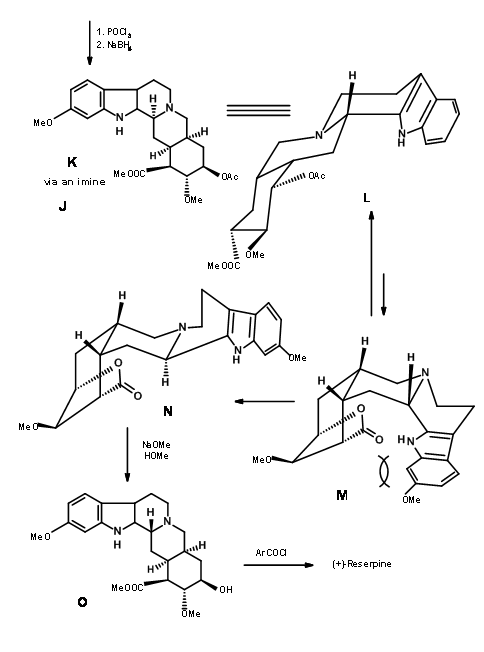
Completion of the A, B, C, D and E rings: Condensation of 9.2I with o-methoxytryptamine followed by Pictet-Spengler condensation led to the formation of Isoreserpine and not Reserpine. Woodward reasoned that this was due to the fact that the C, D and E rings in all-trans geometry, had all substituents in the stable equatorial orientation in Isoreserpine. Woodward executed the isomerisation at C3 in an ingenious way. Aqueous alkali hydrolyzed the acetate – ester functions, which was than lactonised under DCC conditions. Thus, an unstable all-axial E ring was locked in as a lactone (9.2M). This forced the molecule in a crowded unstable state. On acid catalyzed isomerisation with the high boiling carboxylic acid, the C3 position isomerized to the reserpine configuration. On transesterification, the correct isomer was formed with a free –OH ready for final acylation.
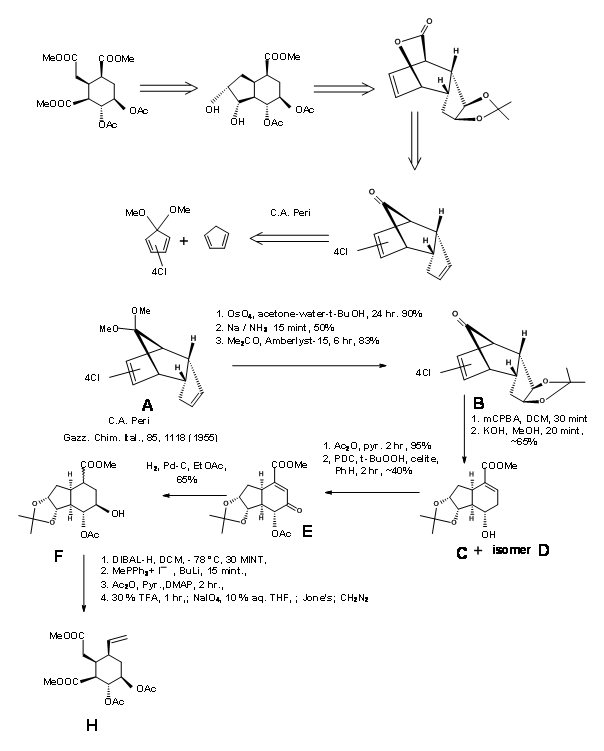
G. Metha’s Synthesis of E ring: G. Metha et.al., (J. Chem. Soc., Perkin Trans. 1, 1319 (2000)) controlled the stereochemistry on the E ring through a bicyclo[2.2.1]heptane system as shown in Figure 9.3.
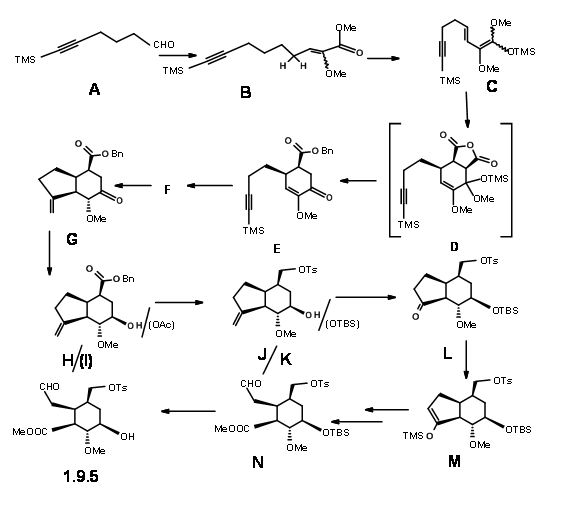
Stork’s synthesis: Gilbert Stork (J. Am. Chem. Soc., 127, 16255 (2005) reasoned that the stereochemistry at C3 in the (AB ABDE ABCDE) cyclisation route of Woodward could be controlled if an iminium intermediate 9.4 could be formed first. This would then orient a chair-like folding of the tether chain (potential C ring) for an axial attack on the iminium ion to give a stereoselective C/D ring closure to form reserpine. They reasoned that the intermediate 9.5 would serve as the key intermediate.

9.4 (left) and 9.5 (right)
After some unsuccessful attempts, Stork’s school completed the synthesis of 9.5 using a route shown in Figure 9.6. Condensation of the hexynal 9.6A with the lithium enolate of methyl methoxy acetate followed by benzenesulphonyl chloride and further heating with DBU gave the conjugated methoxy ester with the (Z) isomer 9.6B as the dominant product. Trimethylsilyl chloride trapped the diene ketene acetal 9.6C without purification of the intermediate product. D.A reaction with maleic anhydride followed with aqueous THF gave the decarboxylated acid that was esterified to give 9.6E. A free-radical cyclisation with tributylstannane in refluxing t-butanol and subsequent workup led to an epimeric mixture that was equilibrated with base to the desired isomer 9.6G. Reduction of the keto- group with L-Selectride gave the axial alcohol. This alcohol was the inverted via mesylation and treatment with cesium acetate to give 9.6I. Reduction with LAH, selective tosylation of the primary alcohol and silylation of the secondary alcohol and ozonolysis gave 9.6L via 9.6K. Opening of the 5-membered ring was achieved by trapping the kinetic enolate as TMS derivative 9.6M and ozonolysis and final esterification gave 9.6N. This key intermediate was also synthesized via an alternate more efficient route.
Unfortunately, condensation of 9.5 with the indole precursor gave isoreserpine as the major product (Fig 9.7). The researchers reasoned that this unexpected result was most probably due to formation of the
C ring prior to cyclisation. In order to drive the reaction towards a DE ring formation, they decided to trap the intermediate imine as the cyanoamine 9.8A. This was achieved by adding an excess of potassium cyanide to the reaction mixture
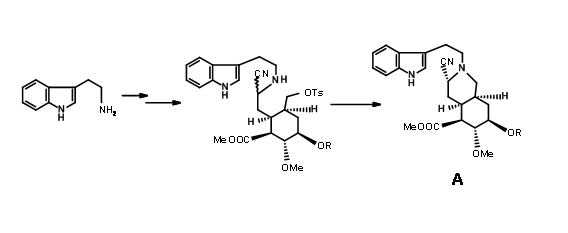
The expected product 9.8A with an axial cyano- group was the sole product. Refluxing the aminonitrile in acetonitril as solvent again gave (±) methyl isoreserpine precursor as the major product (Fig 9.9).Since the expected chair-like folding and an axial attack of the indole moiety on the iminium ion moiety appeared to be on sound srereoelectronic grounds, the authors reasoned that the cyanide anion on the α- phase of the molecule formed a tight ion pair with the immonium ion
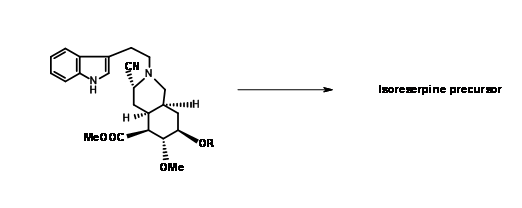
under these conditions.This anion prevented axial attack on a chair-like folding of the chain, forcing a boat like conformation prior to cyclisation followed by an axial attack to give isoreserpine as a major product.
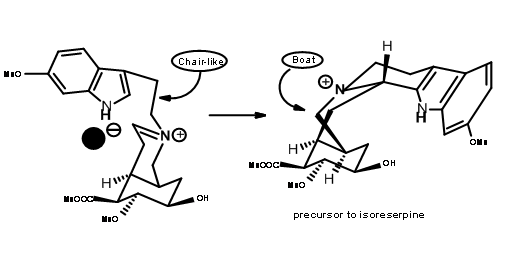
In such an event, allowing the cyanide counter ion to escape from the influence of the iminium ion by the use of a polar solvent should clean the reaction site for a chair-axial attack. When the nitrilamine 9.8A was treated 10%
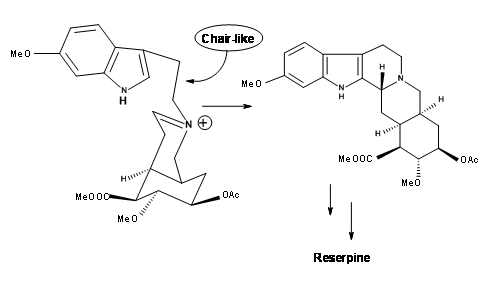
solution of 1 N HCl in THF solvent, the precursor for (±) methyl reserpine was the product in 90% yield. This was eventually converted to reserpine by known procedures. Based on these results, another very short chiral route has been reported by the authors. Thus, persistence and critical mechanistic reasoning at every point of failure led to a successful direct synthesis of the correct isomer.
S. Hanessean et.al., (J.Org. Chem.,62, 465 (1997) applied the Chiron approach to the key intermediate 9.10B suitable for the CD ring formation in a E ABE ABCDE approach discussed so far.
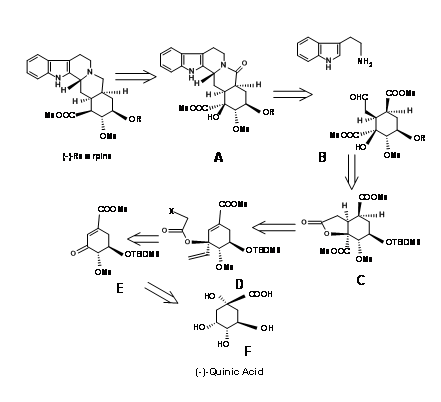
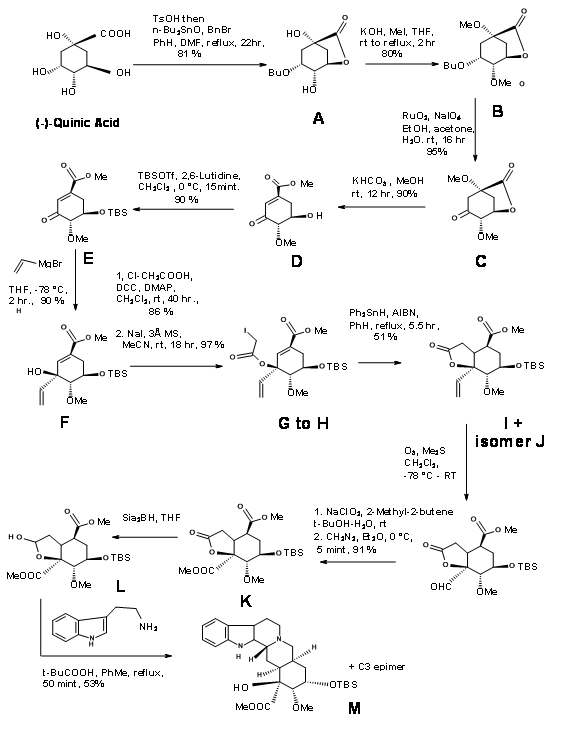
Quinic acid 9.10F had most of the chiral centers in the proper orientation. However it needed modification at the potential C20, C15 and C16 centres. Retroanalysis of Hanessian is shown in Figure 9.10. The actual synthesis is shown in Figure 9.11. Lactonisation, followed by selective benzyl protection and methylation of the remaining OH groups gave the compound B. Oxidation was followed by methanolysis of lactone ring led to elimination to give the conjugaten ketone D. Protection of the C18 – OH as TBDMS followed by vinylation using Grignard reagent placed the potential carboxylic acid function. The t-alcohol thus generated provided an anchor the stereospecific introduction of the needed two carbon chain. A reductive free radical cyclisation placed the carbon chain in the correct stereochemistry. The correct isomer at C20 (I) was then moved to the next step. This key intermediate L led to the reserpine and isoreserpine precursors 9.11M and 9.11M’ in the ratio 1.4 : 1 respectively. These steps are shown in Figure 9.11.