
6: Strategies in (-)-Menthol Synthesis
- Page ID
- 21840

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)(-)-Menthol is amongst the most important perfume / flavor chemical, extensively used in pharmaceuticals, cosmetics, toothpastes, chewing gums and toiletries. Out a the estimated total production of about 20,000 m.tons, natural menthol accounts to about 13 m.tons, the rest coming from synthetic sources. The natural source - oil of Mentha Arvensis - being erratic due to dependence on monsoon, the demand for synthetic menthol is on the increase. The manufacturing processes chosen for discussions here demonstrate three important methodologies used in industry for the synthesis of chiral compounds. A summary of some of the known processes is provided in Figure 6.1.
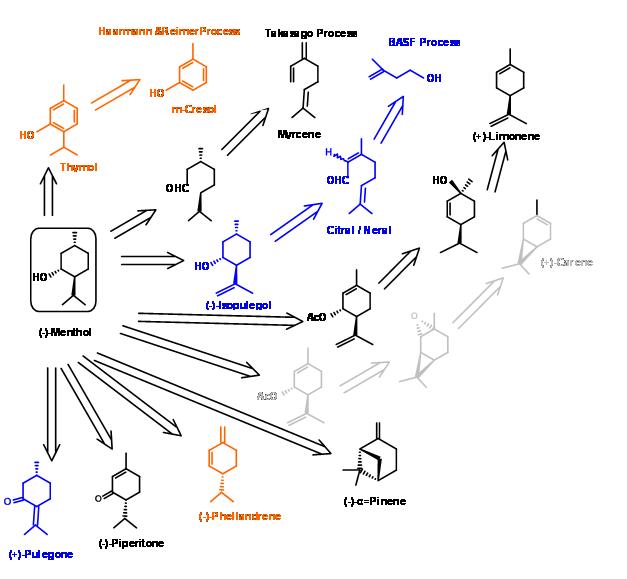
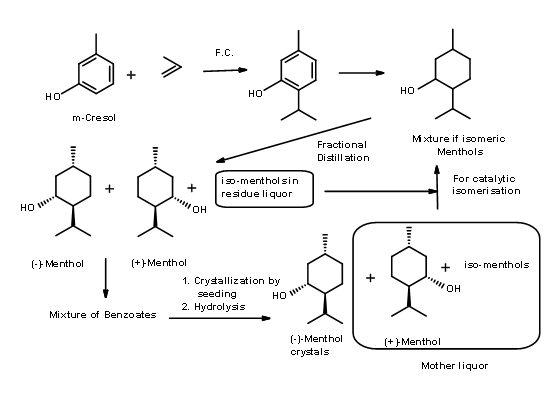
Symrise Process (formerly know as Haarmann & Reimer process) (US Patent 3,943,181 (Mar 9 1976)) – In this process (Fig 6.2), thymol is synthesized from m-cresol. Catalytic hydrogenation gave a mixture of Menthols from which menthols were first obtained as a racemic mixture by careful fractional distillation. The residual mixture was epimerised to increase the content of racemic menthol using a patented catalytic process. The breakthrough in the process is the resolution of the benzoate ester of the racemate by recrystallization by a process of seeding the concentrate with one pure epimer. The mother liquor that was now rich in the (+) isomer was recycled by taking it back to the distillation cycle. In this process, overall yield of (-)-menthol is about 90%.
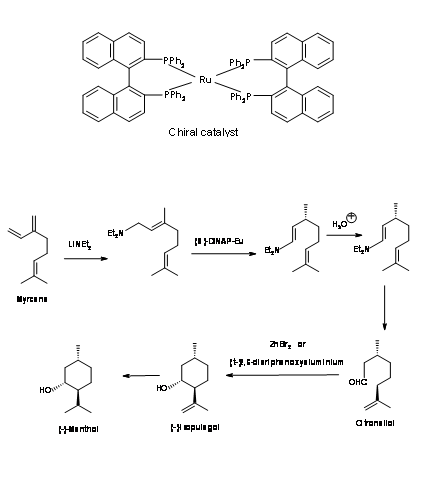
Takasago Process: In this process a (S)-DINAP catalysed isomerization is the key step Fig 6.3. Addition of lithium amide to Myrcene gave an addition compound that was isomerised using a chiral ruthenium catalyst. Hydrolysis of the resulting enamine gave an aldehyde citronellal in high enantiomeric purity. This was cyclized by Lewis catalyst. Catalytic reduction of the olefin gave (-)-Menthol1.
BASF Process: BASF has already set up processes for the synthesis of a series of terpenes starting from butene. In view of the high demand for (-)-Menthol, they extended the product chain to (-)-Menthol as well. The scheme for the synthesis of the product chain is shown in Figure 6.4.
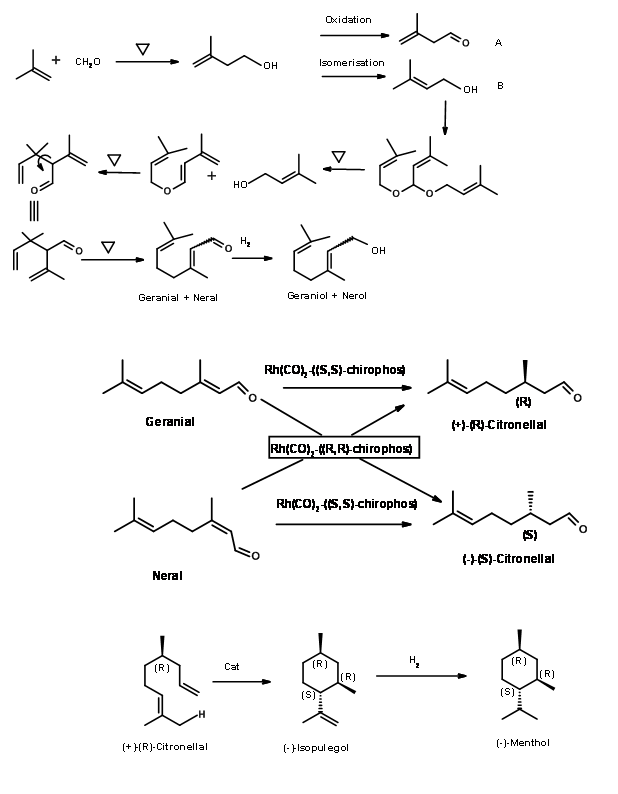
Fig 6.4
Extention of the Citral value chain to (+)-Citronellal, (-)-Isopulegol and finally to (-)-Menthol gave a range of value added products. Note that these processes have taken advantage of the developments in catalytic processes in recent years. Details of the catalysts are not made public.
References
- Preparation of citral, United States Patent 4288636 (09/08/1981)
- Method for the production of menthol, United States Patent 7709688 (05/04/2010);
- Enantioselective hydrogenation of allylic and homoallylic alcohols, J. Am. Chem. Soc., 1987, 109 (5), pp 1596–1597
- Norbert; Method for the production of isopulegol, United States Patent 7550633 (06/23/2009);
- Diarylphenoxy aluminum compounds, United States Patent 7608742 (10/27/2009)
- CONTINUOUS PROCESS FOR PREPARING MENTHOL IN PURE OR ENRICHED FORM, WIPO Patent Application WO/2009/033870 (03/19/2009).
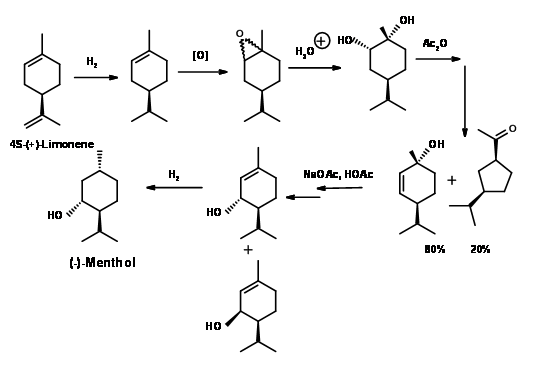
(-)-Menthol from (+)-Limonene(Fig 6.5): Limonene is abundantly available from peels of citrus fruits. On selective catalytic reduction with Ra-Ni, it could be reduced to (+)-1-Menthene, which on epoxidation and hydrolysis gave (+)-1-hydroxyneocarvomenthol. Acylation followed by pyrolysis gave (-)-trans-menth-2-ene-1-ol as the major product. The crude product was solvolysed to give a mixture of piperityl actates as the allylic migration products. The crude product was distilled at this stage to separate the cis- and trans piperitols. The minor ring contraction product was useful as perfume intermediate elsewhere. The final reduction was achieved by H2 / Pd-C to give 75% yield of (-)-Menthol after fractional distillation.
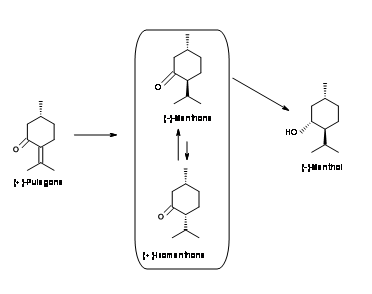
(-)-Menthol from (+)-Pulegone(Fig 6.6): The Starting material has the correct configuration at C1. The problem is to reduce the double bond enantioselectively. The double bond is first reduced by catalytic hydrogenation to give a mixture of (-)-Menthone and (+)-Isomenthone. The all-equatorial configuration of (-)-menthol is best attained by dissolving metal reduction. The enolate intermediate is protonated to the thermodynamically stable (-)-menthol.
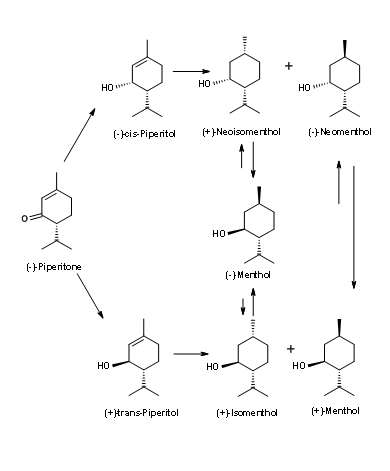
(-)-Menthol from (-)-Piperitone (Fig 6.7): Look closely at the stereochemistry at the asymmetric center of (-)-Piperitone. The isopropyl is in the wrong configuration for (-)-Menthone. The challenge here is (1) isomerise this center and (2) enantioselectively reduce the double bond. All attempts to produce (-)-Menthol produce only mixtures as shown in Figure 6.7. Hence this process has not been very successful.
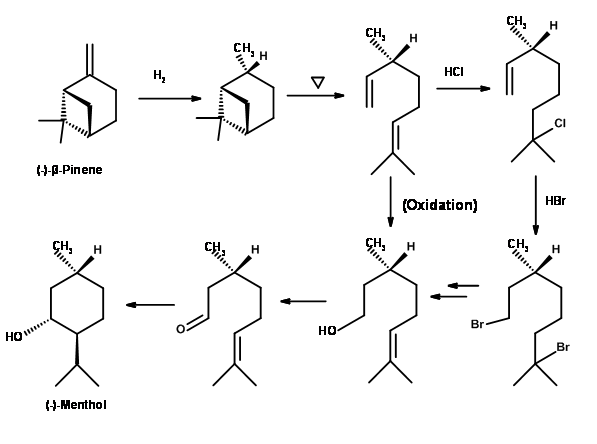
(-)-Menthol from (-)-β-Pinene (Fig 6.8):(-)-β-Pinene offers a good route because the requisite structural features are present and is available in sufficient optical purity. Hydrogenation of (-)-β-Pinene gave cis-Pinane is a major product. On pyrolysis, the strained bridged ring system cleaved to give optically pure 2,6-dimethyl-2,7-octadiene. It was converted to (+)-Citronellol in good yields by direct oxidation. Alternately the more substituted olefin was first subjected to a Markovnikov addition of HCl followed by an anti-Markovnikov addition of HBr. Solvolysis reaction provided a mixture of citronellols. Catalytic oxidation of the alcohol provided (+)-Citronellal. This could be converted to (-)-Menthol by known procedures. The product was however contaminated with trace amounts of (+)-Menthol arising from trans-Pinane generated in the first step.
(-)-Menthol from (+)-δ-3-Carene (Fig 6.9): δ-3-Carene is another chiral synthon that has the required structural features to serve as a starting material for (-)-Menthol. Catalytic isomerization of δ-3-Carene gave (+)-δ-2-Carene. Two different routes were investigated. In the first route (+)-δ-2-Carene was pyrolysed to cleave the cyclopropane ring. The resultant diene had the right stereochemistry at C1 and C4. The latter did not matter because this asymmetry is lost soon and regenerated in the process. Treatment of the unconjugated diene with HCl and dehydrohalogenation led to a conjugated diene. Addition of HCl led to an allyl chloride. Solvolysis with acetic acid-sodium acetate provided an SN2’ displacement causing an allylic rearrangement. The resulting piperitol acetates gave (+)-cis and (-)-trans-piperitols which could be fractionally distilled. Pure(-)-trans-piperitols yielded (-)-menthol on hydrogenation.
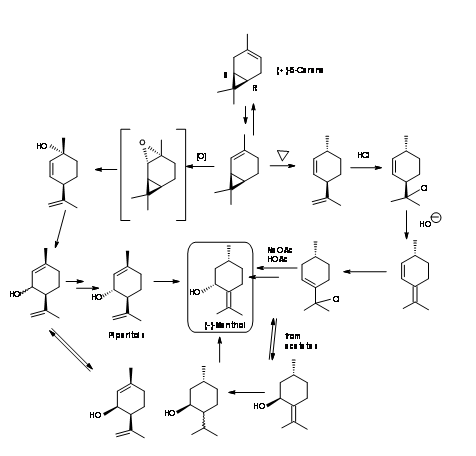
In the second route (+)-δ-2-Carene was epoxidised to yield (+)-cis-2,8-p-menthadienol directly. On buffering with formic acid – acetic acid mixture, allylic rearrangement occurred to give a mixture of formate and acetate of piperityl esters. The corresponding alcohols could be fractionally distilled. The cis-isomer could be isomerised to improve the yield of pure (-)-trans-piperitol. Hydrgenation gave (-)-Menthol.
Conclusion: In this section we were focused on development of commercially viable routes for (-)-Menthol. Note the way modern reagents have influenced industrial processes.
References
- Leffingwell, J.C. & R.E. Shackelford, Laevo-Menthol - Syntheses and organoleptic properties, Cosmetics and Perfumery, 89(6), 69-89, 1974
- Hopp, R., Menthol: its origins, chemistry, physiology and toxicological properties, Rec. Adv. Tobacco Science, Vol. 19, 3-46 (1993).