
5.3: CHEM ATLAS_3
- Page ID
- 408787

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)How This Connects: Unit 3, Lectures 21-30
The purpose of this document is to serve as a guide and resource that gives you a quick overview of each lecture. For each lecture, there is a summary of the main topics covered, the Why This Matters moment, and the new Why This Employs section, plus a few example problems. So why did we make this? We hope it’s useful to get a good snapshot of any given lecture. Whether you couldn’t make it to a lecture or you couldn’t stop thinking about a lecture, this is a way to quickly get a sense of the content. It also gives me a chance to provide additional details that I may not have time for in the Why This Matters example, and also it lets me try out the Why This Employs section, which I certainly will not have time to discuss much in the lecture. Hopefully you find it useful!
One point about these lecture summaries. Please note that the lecture summaries are not meant to be a substitute for lecture notes. If you were to only read these summaries and not go to lecture, yes you’d get a good sense of the lecture from a very high level view, but no, you wouldn’t get enough out of it for it to be your only resource to learn the material!
Below is an image of the Exam 3 Concept Map. This demonstrates how each of the aspects of the course fit together: you have lots of resources! The Practice Problems, Recitations, Goodie Bags, and Lectures are ungraded resources to help you prepare for the quizzes and exams. All of the material listed on this concept map is fair game for Exam 3.

Lecture 21: Bragg’s Law and x-ray Diffraction
Summary
X-ray diffraction (XRD) is a method used for characterizing solids. It relies on the diffraction of x-rays upon striking crystal planes (the Miller planes we’ve learned about!) By assuming that each plane of atoms is continuous, and that they reflect the incoming x-rays such that the incident angle and the reflected angle are equal, the Braggs derived the equation that bears their name and relates the distance between repeating planes (\(\mathrm{d}\)) and the x-ray angle of incidence (\(\theta\)) to the x-ray wavelength:

Two x-rays striking equivalent Miller planes with the same angle of incidence will constructively interfere if the additional distance that one of the travels is equal to the wavelength of the x-ray. Quantitatively, if \(\lambda=2 d \sin \theta\), the intensity of the outgoing x-rays with wavelength \(\lambda\) are enhanced. the constructive interference will occur whenever the path length distance is an integer multiple of the wavelength: \(2 d \sin \theta=n \lambda\) for integer \(\mathrm{n}\). For \(3.091\), we'll assume \(n=1\). When constructive interference occurs, a signal will reach the detector in the XRD machine and a peak will be observed in a plot of the x-ray intensity. For destructive interference, no peak will be observed. Knowing the angle that gives rise to a peak as well as the wavelength of the incident x-rays allows us to obtain the distance between the planes that produced the reflection. This is known as the Bragg condition:
\(2 d_{h k l} \sin \theta_{h k l}=\lambda\)
For a given Miller plane, denoted by \((h k l)\), the Bragg condition is satisfied by a pair \((d, \theta)\) of inter-planar spacing and incident angle.

For each of the crystal structures (SC, BCC, or FCC), there are reflections that even when the Bragg Condition is met lead to destructive interference, due to crystal symmetry. The pattern of peak absences was used to derive a set of rules called selection rules, which allow us to know, or at least narrow down the possibilities of, the crystal structure of a material based on its XRD peaks. For the case of SC, there are no rules and any plane is fine. There are no forbidden reflections.
For the case of \(\mathrm{BCC}\), allowed reflections are those where \(\mathrm{h}+\mathrm{k}+\mathrm{l}\) is an even number. Forbidden reflection are those for which \(\mathrm{h}+\mathrm{k}+\mathrm{l}\) is an odd number. For the case of \(\mathrm{FCC}\), allowed reflections are those where \(\mathrm{h}, \mathrm{k}, \mathrm{l}\) are all odd or \(\mathrm{h}, \mathrm{k}, \mathrm{l}\) are all even. Forbidden reflections of FCC are those where h,k,l is mixed odd/even.

Why this matters
For solids, structure can be as important as the chemistry itself, and they are deeply connected. When I look up the crystal structure of a element in the Periodic Table I see what it is for the ground or lowest energy state of that element. This is the overall “happy place,” energetically speaking, of the material. But materials can take on other, metastable structures and be very happy there, too. And Why This Matters is because the properties can be completely different depending on which crystal structure the material takes, and XRD is the single most important characterization method we have to determine crystal structure. We’ve already seen the difference between graphite and diamond, which contains the same exact element (carbon) but just arranged in a different structure. The same is true for, well, pretty much everything. Take another element, iron, as an example.

Here’s a phase diagram for iron. As you may remember from Lecture 14, a phase diagram is a plot of the different phases of a material as a function of some variables, in this case pressure and temperature. Notice that at normal, or “ambient,” temperature (\(\approx 300\mathrm{K}\)) and pressure (\(\approx 1 \mathrm{bar}\)) conditions, iron is a \(\mathrm{BCC}\) crystal. This is also what you’ll find if you look up its crystal structure in the PT. But notice from reading the phase diagram that if we raise the temperature it becomes \(\mathrm{FCC}\), and if we raise the pressure it goes into the HCP (it’s not cubic so we haven’t covered it) phase. Fun fact: if you keep raising the temperature eventually it will go back to being \(\mathrm{BCC}\).
The reason all of this matters is that the structure changes the properties. In the case of iron, the element’s magnetic properties are affected. If it’s FCC then it will be ’antiferromagnetic’ as opposed to the BCC ’ferromagnetic’ behavior. I recommend taking a magnetics course in the future to learn more about these terms! This has huge implications in magnetic technologies. And I know you’re probably thinking: sure, but we don’t build too many iron-based technologies that operate at 1000\(\mathrm{K}\)! You’re right, but the trick is that often times we can coax these materials to get stuck in one of those metastable phases and then use it for technologies while it’s in that phase. Again, diamond is a great example: it’s not the ground state of carbon so it is metastable as diamond instead of graphite, but we know that it stays stuck there for a long time so for most technologies that use diamond (say, a piece of jewelry), we don’t worry about it changing out of its metastable phase.
Coming back to the topic of the lecture, when we make something, whether that something is as old as elemental iron or as new as a nanostructured perovskite, the simplest and most common way we have to tell its crystal structure is by \(\mathrm{XRD}\). In some cases, the use of \(\mathrm{XRD}\) can unravel the structural mystery of a material, as in the case of the double-helix for DNA or the many proteins since. In other cases, it’s used to not unlock the secret to a completely new structure, but rather to classify a material into one or the other well-known structures. Sometimes the reason is to understand a material, sometimes it’s to engineer the material properties, and often times it’s both. But whatever the motivation, this incredibly powerful characterization tool has revolutionized what we know about solids.
Why this employs
We’ve been referencing these crystallographers (who are very picky about notation!) for several lectures now. But who are these people? And more to the point: who hires them? A whole lot of X-ray crystallographer jobs are out there in the biotech industry, in companies of all sizes. Blueprint Medicines, which looks like a Harvard spin-out and is just down the street, has an opening now with the title, “Senior Scientist/Principal Scientist, X-Ray Crystallography,” with the first job function description being, “Provide x-ray crystallographic and protein structure-function support, including structure-based drug design, to on-going drug discovery projects and new target discovery initiatives.” And the larger pharmaceutical and biotech companies have even more jobs. Take Novartis, which also has a big headquarters near MIT, just down Mass Ave. They’re hiring people to perform ”crystallography experiments including crystallization screening using automated liquid handling. GlaxoSmithKline has an opening for someone to “enable higher throughput x-ray crystallography.” Johnson and Johnson is hiring people with X-ray crystallography expertise to do, “automated chemistry.” And I have to mention one last example because of the title of their current opening, Bristol-Myers-Squibb is looking for a, “Research Investigator, Solid-State Chemistry.” Gotta love it! They want, “an entry level scientist with background in X-ray crystallography, X-ray diffraction, and solid-state characterization.” That’s now you!
And it’s not all about pharma. Hospitals are hiring X-ray crystallographers too (these are not the same position as a radiologist), to work on research projects for example with openings at Mass General and Dana Farber. And many research positions in X-ray diffraction are out there too, from positions at the Howard Hughs Medical Institute to university labs and centers all across the country.
Example Problems
1. Determine the structure (simple cubic, body centered cubic, or face centered cubic) to which this \(\mathrm{XRD}\) pattern most likely corresponds (copper \(\kappa_{\alpha}\) x-rays were used).

- Answer
-

Given that the indices of each plane are either all odd or all even, using the selection rules we are able to determine that this structure is \(\mathrm{FCC}\).
Lecture 22: From x-ray Diffraction to Crystal Structure
Summary
This lecture we finished analyzing the \(\mathrm{XRD}\) spectrum of an \(\mathrm{Al}\) sample, shown below.

The plot was obtained by shining \(\mathrm{K}\)-alpha x-rays from a \(\mathrm{Cu}\) target onto our \(\mathrm{Al}\) sample. What we want to do is figure out the crystal structure and the lattice constant of \(\mathrm{Al}\). To answer these questions, we need our handy Miller plane separating distance equation (where ' \(\mathrm{d}\) ' is the distance between two repeating Miller planes with indices \(\mathrm{hkl}\) in a cubic system, and ' \(a\) ' is the lattice constant):
\(d_{h k l}=\dfrac{a}{\sqrt{h^2+k^2+{ }^2}}\)
and the Bragg condition:
\(2 d_{h k l} \sin \theta_{h k l}=\lambda\)
Notice that both of these equations include \(d_{h k l}\). We can use this to our advantage and substitute one equation for \(d_{h k l}\) into another to obtain the following:
\(\left(\dfrac{\lambda}{2 a}\right)^2=\dfrac{\left(\sin \theta_{h k l}\right)^2}{h^2+k^2+l^2}\)
We know the value of the wavelength, because it is fixed by the \(K_a\) x-rays from the copper source. These x-rays have a wavelength of \(1.54 \AA\). So the expression for our example becomes:
\(\left(\dfrac{1.54 \AA}{2 a}\right)^2=\dfrac{\left(\sin \theta_{h k l}\right)^2}{h^2+k^2+l^2}\)
Now we have constants on both sides of the equal sign, because the lattice parameter does not change. We can make an educated guess of the \(\mathrm{hkl}\) value by following the procedures outlined in the chart on the following page.
From the selection rules we know that \(\mathrm{Al}\) is an \(\mathrm{FCC}\) metal, since the (\(\mathrm{hkl}\)) combinations are always either all even or all odd. We can also take a value of \(\theta\) and a value of h, k, and l and plug these into the equation above to find the lattice parameter, given in the rightmost column of the chart.

Why this matters

Photo of Henry Moseley is in the public domain.
This is Henry Moseley (image, Royal Society of Chemistry). He’s pictured there in his lab, holding in his hands, of course, a modified cathode ray tube. He was experimenting with X-rays. But Moseley’s interests were less about the crystal structures and Bragg conditions and more about the X-ray lines themselves and what they meant. He carried out a systematic study of the metals used to generate the Xrays, comparing the X-ray emission from 38 different chemical elements.
Some work had already been done that led to our understanding of the characteristic and continuous parts of the X-ray generation spectrum, as we discussed a few lectures ago. But a full systematic study had not been carried out until Moseley’s work. Take a look at the difference between the two X-ray spectra generated with two different targets: \(\mathrm{Mo}\) and \(\mathrm{Cu}\). Note the \(\mathrm{K}_{\alpha}\) and \(\mathrm{K}_{\beta}\) lines for each one, and that they’re shifted to lower wavelength for \(\mathrm{Mo}\) compared to \(\mathrm{Cu}\). As we know, this is because of difference in energy between the shells of \(n=1\) and \(n=2\) (for \(\mathrm{K}_{\alpha}\)) or \(n=1\) and \(n=3\) (for \(\mathrm{K}_{\beta}\)), and it makes sense this energy difference is greater (corresponding to lower wavelength) for \(\mathrm{Mo}\) since it’s heavier than \(\mathrm{Cu}\). But what exactly is the dependence, and why is it present? Let’s look at the data for a sequence of targets, directly from a subset of Moseley’s data.

These are characteristic X-ray lines for \(\mathrm{Ca}\) up through \(\mathrm{Zn}\), so going across the \(\mathrm{d}\)-block elements of the fourth row in the PT. Note that it’s not actually \(\mathrm{Zn}\) but rather brass, which we’ve already learned is a mixture of \(\mathrm{Zn}\) with \(\mathrm{Cu}\) – that’s because \(\mathrm{Zn}\) would melt under the high energy electron bombardment before it could give off any characteristic X-rays, so Moseley gave it the extra strength it needed by making brass, and then subtracted out the emission from \(\mathrm{Cu}\). Nice trick!
What Moseley found was that if the characteristic emission lines were plotted as the square of their energy vs. atomic number, that you'd get a straight line. He fit the data by considering the lines to come from a core excitation, so a difference in energy levels from Bohr's model:
\(E_{x-r a y}=13.6[e V](Z-1)^2\left(\dfrac{1}{1^2}-\dfrac{1}{2^2}\right)=\dfrac{3}{4}(13.6[\mathrm{eV}])(Z-1)^2\)
This is now called Moseley's law. Can you see why it's \(\mathrm{Z}-1\) instead of \(\mathrm{Z}\) as in the Bohr model? It's because the electron cascading down to generate the X-ray is "seeing" a 1-electron screening of the nucleus. That's because one of those core 1s electrons was knocked out, but there's one left there that screens out a positive charge, hence the \(\mathrm{Z}-1\). So what did this trend in the data mean? Here's what Moseley said in his 1913 paper, "We have here a proof that there is in the atom a fundamental quantity, which increases by regular steps as one passes from one element to the next. This quantity can only be the charge on the central positive nucleus, of the existence of which we already have definite proof."
The reason this is such a big deal, and why I'm making it the Why This Matters for this chapter, is that even through the Mendeleev periodic table had been around and more elements were being discovered and added, there was a major flaw in the periodic table: the position predicted by an element's atomic weight did not always match the position predicted by its chemical properties. Remember that the positioning by Mendeleev was based on weight and properties and when the periodicity called for it, he chose to order the elements based on their properties, rather than their atomic weight. But was there something more fundamental than atomic weight?
Moseley's data only made sense if the positive charge in nucleus increased by exactly one unit as you go from one element to the next in the PT. In other words, he discovered that an element's atomic number is identical to how many protons it has! I know this seems kind of obvious to us now, but back then "atomic number" was simply a number with no meaning, other than the element's place in the periodic table. The atomic number was not thought to be associated with any measurable physical quantity. For Mendeleev, periodicity was by atomic mass and chemical properties; for Moseley, it was by atomic number. This led to a much deeper understanding of the periodic table and his insights immediately helped to understand some key mysteries, for example where to place the lanthanides in the PT \((\mathrm{La}=\# 57, \mathrm{Lu}=\# 71)\), or why \(\mathrm{Co}\) comes before \(\mathrm{Ni}\). And the gaps that Mendeleev brilliantly left open in his PT to create periodicity now made sense by missing atomic numbers in a sequence, for example elements \(43,61,72\), and \(75\) were now understood to contain that many protons (they were discovered later by other scientists: technetium, promethium, hafnium and rhenium).
Moseley died tragically in 1915 at age 27 in a battle in WW1. In 1916 no Nobel Prizes were awarded in physics or chemistry, which is thought to have been done to honor Moseley, who surely deserved one.
Why this employs
We've covered X-ray generation and machine manufacturers two chapters ago, and in the last chapter we looked at jobs related to X-ray crystallography. For this last Why This Employs related to Xrays, it's time to go big or go home. And when I say big, I mean really big. When electrons or for that matter any charged particles are accelerated to near light speeds, then the acceleration they experience simply to stay in a loop produces massively energetic radiation. The wavelength can vary dramatically, but very often these enormous accelerators are used to make super-high-energy X-rays. The intensity of these rays is dazzlingly bright, millions of times brighter than sunlight and thousands of times more intense than X-rays produced in other ways. This level of brightness makes them useful for pretty much any and all areas of research and fields of science. Some types of measurements are only possible when synchrotron light is used, and for other types one can get better quality information in less time than with traditional light sources. They've been shown to be useful in so many areas it's impossible to list them all, but certainly in biology, chemistry, physics, materials, medicine, drug discovery, and geology, to name only a few fields, synchrotrons have made a dramatic impact. They're in such demand that often one needs to book time on them many months in advance. A typical synchrotron can have as many as 50 "beam lines" that grab the high energy X-rays out of the loop and focus them in a beam where experiments are done. These lines are usually booked and put to use 24 hours per day, 7 days per week, all year round. Here’s a photo of one of them, the Advanced Photon Source at Argonne National Laboratory in Illinois. Their tagline overview statement reads, “The Advanced Photon Source (APS) at the U.S. Department of Energy’s Argonne National Laboratory provides ultra-bright, high-energy storage ring-generated x-ray beams for research in almost all scientific disciplines.”

The Employment part of this is pretty cool. These facilities, which are called “synchrotrons,” are all over the world, and they require thousands of people to build and then run. The international nature of them is astounding: just do a image search for synchrotron and you’ll find pictures of them all over the planet. And that means jobs in many different locations. Some are old, like the one at Berkeley National Lab (but it’s still kicking!), some are medium-sized like that APS pictured above, and some are huge like the Hadron Collider I referenced in the last lecture as a place where 3D X-ray imaging was invented. Colliders are also synchrotron light sources, since they’re built to accelerate particles at very high speeds. Even though colliders may be used to smash particles together at these speeds, they’re also often used simply as a way to generate high intensity light.
I don’t have a specific job title in mind, but if you look at a list like this one: https://en.wikipedia.org/wiki/List_of_synchrotron_radiation_facilities you’ll see where these synchrotrons are, and for each one of them there’s an “employment” link you can click on to explore possible jobs.
Extra practice
1. Determine the element that made up the sample from Lecture 21 Extra Practice Problem 1. The XRD pattern is reproduced below (copper \(k_a\) x-rays were used).

- Answer
-
To know which element was used as the sample, find the lattice parameter (a) using the equation for interplanar spacing \(\left(d_{h k l}\right)\), Bragg's law, and Moseley's law. For \(\mathrm{h,k,l}\) and \(\theta_{h k l}\), pick a plane and a corresponding angle from the chart we developed last chapter, shown below:

\begin{gathered}
d_{h k l}=\dfrac{a}{\sqrt{h^2+k^2+l^2}} \\
\lambda=2 d_{h k l} \sin \left(\theta_{h k l}\right)
\end{gathered}The energy corresponds to \(\mathrm{Cu}(\mathrm{Z}=29) k_\alpha\) radiation:
\begin{gathered}
E=\dfrac{h c}{\lambda}=13.6[e V](Z-1)^2\left(\dfrac{1}{n_f^2}-\dfrac{1}{n_i^2}\right) \\
\lambda=\dfrac{h c}{-13.6(Z-1)^2\left(\frac{1}{n_f^2}-\frac{1}{n_i^2}\right)} \\
d_{h k l}=\dfrac{h c}{-13.6(Z-1)^2\left(\frac{1}{n_f^2}-\frac{1}{n_i^2}\right) 2 \sin \theta_{h k l}} \\
a=\dfrac{h c \sqrt{h^2+k^2+l^2}}{-13.6(Z-1)^2\left(\frac{1}{n_f^2}-\frac{1}{n_i^2}\right) 2 \sin \theta_{h k l}}=3.53 A
\end{gathered}This lattice parameter corresponds to \(\mathrm{Ni}\).
2. You would like to perform an XRD experiment, but you don't know what target is used in the diffractometer in your lab. You put in a calibration sample of iron, which is BCC and has a lattice parameter of \(2.856\) angstroms. If you observe the following XRD pattern, what material is the target? You are pretty sure that there is a filter that prevents anything with lower energy than \(k_a\) radiation from hitting your sample.
The peaks observed are as follows:
| counts | 10 | 1000 | 20 | 2200 | 8 | 5 | 1200 | 2500 |
|---|---|---|---|---|---|---|---|---|
| \(2\theta\) | 17.38 | 20.87 | 24.67 | 29.62 | 30.30 | 35.15 | 36.45 | 42.37 |
a) What kind(s) of x-rays are hitting the sample?
- Answer
-
\(k_\alpha \operatorname{AND} k_\beta\)
b) How many planes are represented by the data? Which planes are they?
- Answer
-
4 planes are represented: \((110),(200),(211)\), and \((220)\)
c) What are the interplanar spacings associated with these planes?
- Answer
-
\[\dfrac{2.856}{\sqrt{h^2+k^2+l^2}} \nonumber\]
Plugging in each (\(\mathrm{hkl}\)), the spacings are 2.02, 1.43, 1.17, and 1.01 A
d) Which element was used as the target?
- Answer
-
\begin{gathered}
\lambda=2 d_{h k l} \sin \left(\theta_{h k l}\right) \\
\lambda_{k_\alpha}=0.73 A \\
\lambda_{k_\beta}=0.61 A
\end{gathered}
- Answer
-
\[E=\dfrac{h c}{\lambda}=13.6(Z-1)^2\left(\dfrac{1}{n_f^2}-\dfrac{1}{n_i^2}\right) \nonumber\]
For \(k_\alpha, n_i=2\) and \(n_f=1\). For \(k_\beta, n_i=3\) and \(n_f=1\).
\begin{aligned}
&h=4.135 \times 10^{-15} \mathrm{eV} . \mathrm{s} \\
&c=3 x 10^8 \mathrm{~m} / \mathrm{s} \\
&\mathrm{Z}=42
\end{aligned}
Lecture 23: Point Defects
Summary
A point defect is a localized disruption in the regularity of the crystal lattice. There are four types of point defects: vacancies, interstitial impurities, self-interstitials, and substitutional impurities.
Arrhenius determined a law for the temperature dependence of the rate at which processes occur:
\(k=A e^{-E_a / R T}=A e^{-E_a / k_B T}\)
where \(\mathrm{R}\) is the gas constant (or \(k_B\) is the Boltzmann constant) and \(E_a\) is the activation energy. The term in the exponent should be unitless: therefore, if the activation energy is given in \(\mathrm{J} / \mathrm{mol}\), use the version with \(R\), but if the activation energy is given in \(J\), use the \(K_B\) version. Recall that the gas constant is just \(R=K_B * N_A\). The units are determined by the prefactor \(\mathrm{A}\), which can be thought of as an average kinetic energy of the system.
Vacancies are always present in every solid because they're a result of thermally-activated processes. We can consider the rate of formation of a vacancy and the rate of removal of that same vacancy as two thermally-activated processes, each with their own rate. At any given temperature, when the rate of forming the vacancy is the same as the rate of "de-forming" the vacancy then the vacancy concentration in the crystal will be in equilibrium. Since each rate is thermally activated we can use an Arrhenius equation to describe both the forward and back process, and setting them equal for equilibrium one arrives at a formula describing how the vacancy concentration depends on temperature:
\(N_v=N e^{-E_a / k_B T}\)
where \(N_v / N\) is the fractional concentration of vacancies and \(E_a\) is the activation energy \([\mathrm{J}]\) required to remove one atom. If the vacancy occurs in an ionic solid, charge neutrality must be maintained. Therefore, the defect either forms as a Schottky defect, where a pair of charges (one cation and one anion) is removed, or as a Frenkel defect, where the vacant atom sits elsewhere in the lattice on an interstitial site. For a Schottky defect in an ionic solid like \(\mathrm{CaCl}_2\), two anions \(\left(\mathrm{Cl}^{-}\right)\) must be removed for each cation \(\left(\mathrm{Ca}_2^{+}\right)\) vacancy to maintain charge neutrality. Frenkel and a smaller cation, like \(\mathrm{AgCl}, \mathrm{AgBr}\), and \(\mathrm{AgI}\), for example.

Interstitial defects can occur in covalent solids as well: in this case, an extra atom occupies a site that is not part of the lattice, but the charge neutrality requirement doesn't necessitate the creation of a vacancy provided the interstitial atoms have the same charge as the lattice atoms. For example, a \(\mathrm{C}\) interstitial in \(\mathrm{Fe}\) is charge neutral. If the interstitial atom is the same type of atom as the lattice, like a \(\mathrm{Si}\) atom in a \(\mathrm{Si}\) lattice but not on a lattice site, the defect is called a self-interstitial. The energy required to form a self-interstitial \((2-5 \mathrm{eV})\) is much higher than for a vacancy \((0.5-1 \mathrm{eV})\), so these defects are much less common: this can be rationalized by thinking about how hard it would be to squeeze an atom between similarly-sized atoms arranged in a closely-packed lattice.
Atoms which take the place of another atom in a lattice are called substitutional defects. Generally, the Hume-Rothery rules provide guidelines to which atoms can be a substitutional defect: the atomic size must be within \(+/- 15\%\), the crystal structure must be the same, the electronegativity must be similar, and the valence must be the same or higher.

Why this matters
Let’s pick up on the interstitial defect of carbon in iron, otherwise known as steel. This particular defect is one that has positive benefits if it’s controlled carefully and the right amount of carbon (not much, it turns out) is placed in the right positions within the iron lattice (the tetrahedral holes, for example, in bcc \(\mathrm{Fe}\)). In fact, the change in iron’s properties are absolutely tremendous and represent a spectacular example of how defects can be used beneficially. If you Production vs. time figure removed due to take a piece of pure iron and ap- copyright restrictions. ply sideways strain on it, then its resolved shear stress is quite low, around 10 MPa. That means that if you push sideways on a piece of pure iron it will deform under 10 MPa of pressure. But with just 1% \(\mathrm{C}\) on interstial sites, the \(\mathrm{C}\)-doped iron can have a resolved shear stress as high as 2000 MPa, 200 times larger than the undoped case!

Now, this phenomenon has been known and practiced for over 2500 years, when people first observed the mechanical strength imparted on iron when it was heated by a charcoal fire (the charcoal was the carbon source). But that’s just it: 2500 years ago, or 1000 years ago, or even just 100 years ago, not a whole lot of steel was being made each year. This is now changing, and it’s changing dramatically, and it’s Why This Matters.

Industrial Carbon Emission Chart
Take a look at the chart above (from US Geological Survey, UN, FAO, World Aluminium Association) of the production amount for some of the materials humans make on a scale massive enough to require enormous chunks of the world’s energy consumption. Cement and steel are the top two, and estimates put them at 10-15% combined of annual global \(\mathrm{CO}_2\) emissions. If we look at just \(\mathrm{CO}_2\) emissions from industrial processes, steel has the biggest share at 25% of the total. But even more important are those slopes in the production trends: note that the use of these products is growing and will continue to grow dramatically into the future. Back 2500 years ago it didn’t matter how steel was made. They also had no idea why the charcoal gave iron those properties. Today, not only do we need to find new ways to make steel more efficiently, but we also know what’s happening in the material at the atomic and bonding scale. In other words, we understand its solid state chemistry.
How can we make steel in a more energy efficient manner? Answering that question relies on knowledge of the point defects in the material, and specifically on the energy it takes to get carbon into the interstitial lattice. And it’s not always obvious. For example, if we compare the atomic size of \(\mathrm{C}\) with the sizes of the available interstitial volumes in \(\mathrm{Fe}\), it’s clear that it doesn’t quite fit, and some type of lattice distortion will have to take place in order to accommodate the interstitial defect, even as small as a \(\mathrm{C}\) atom. But that means it’s not as simple as occupying the defect site with the most room, since we need to know how atoms get strained in response to the defect being there. Take the example of -iron: in that phase you would think a \(\mathrm{C}\) atom occupies the larger tetrahedral hole, but in fact it prefers to go to the octahedral interstitial site. The reason for this preference is that when the \(\mathrm{C}\)-atom goes into the interstitial, strain gets relieved for the octahedral site by two nearest neighbor iron atoms moving a little bit, while for tetrahedral site, four iron atoms are nearest-neighbor and the displacement of all of these requires more strain energy. This is just one phase of iron and two different sites. There may be ways to move \(\mathrm{C}\) atoms into other phases more easily, that would take less energy, and give the same strength. Or perhaps there are other ways beyond fire (which is why steel-making takes so much energy) to get the defect chemistry just right. This is a hard problem, but it’s a critical one: take a look at this chart from a recent paper published in Science that breaks down which sectors will be hardest to make “green.” Note the prominence of steel and cement! To solve such hard problems, we will need advances in defect chemistry.
Why this employs
This is an easy one: there’s actually a job position called “Defect Engineer”! At Global Foundries, they care about defects for the basic manufacture of semiconductor materials while at Intel, there are openings for Defect Engineers to work on 3D XPoint which is a new non-volatile memory technology. Intel also has an opening for a “Defect Reduction Engineer.” These and so many more similar openings are for industries where devices are made in clean rooms, often very clean cleanrooms. A “Class 1” cleanroom, for example, means that if you take a meter cubed of air at any given place in the room, you would count less than 10 particles of size 100 nanometers, less than 2 particles size less than 200 nanometers, and zero particles larger than that. The reason fabrication facilities need such a high level of air purity is that they’re making features on the order of 10’s of nanometers and so any small defect can be a big problem.
Now, then again, these are not necessarily point defects (although certainly 100 nm particles that hit a layer of silicon while its being processed can cause point defects). How about technologies where the point defect is the key part of the technology itself? In that case you may be talking about a solid oxide fuel cell (\(\mathrm{SOFC}\)). These are typically metal-oxides and they work by conducting oxygen ions through the material. But the only way an oxygen ion can move is if there are oxygen vacancies present, and in enough density at reasonable temperatures. Many companies work on developing efficient, low-cost, and low temperature solid oxide fuel cells. Every single car manufacturer for example, has an interest in this as a possible future way to power transportation (Nissan has a cool demo car). Other companies like Precision Combustion, Elcogen, or Bloom Energy all work to build \(\mathrm{SOFCs}\), with that last one stating, “better electrons” *on their homepage. Nice. (although I thought all electrons were identical. . . but anyway). The point is that there’s a growing interest in \(\mathrm{SOFC}\) and an already strong market set to reach $1B by 2024. With greater control over those oxygen vacancies, the ability to make the materials cheaper, and the ability to control defects at lower temperatures, the use of \(\mathrm{SOFC}\) could increase even much more than that!
Extra practice
1. Sketch the following defects:
a) Schottky
b) Frenkel
c) Substitutional impurity+vacancy
d) Self-interstitial
e) Substitutional impurity
f) Vacancy
- Answer
-

2. Solid oxide fuel cells rely on the reaction of fuel with oxygen to form water. A ceramic oxide can be doped to introduce oxygen vacancies that allow charge to conduct through the solid electrolyte. Zirconia \((\mathrm{ZrO} 2)\) can be doped by adding \(\mathrm{Sc}2\mathrm{O}3\). If \(0.5 \mathrm{~g}\) of \(\mathrm{Sc}2\mathrm{O}3\) can be incorporated into \(10 \mathrm{~g}\) of \(\mathrm{ZrO} 2\) while maintaining the zirconia structure, how many oxygen vacancies are generated?
- Answer
-
We are doping \(\mathrm{ZrO}_2\) with \(\mathrm{SC}_2 \mathrm{O}_3\). The \(\mathrm{Zr}^{4+}\) ions are replaced by the \(\mathrm{Sc}^{3+}\) ions, creating a charge imbalance of \(-1\) with each substitution. This means that for every 2 such replacements, or every \(2 S c^{3+}\) ions added, there will be a \(-2\) imbalance, and an oxygen vacancy \(\left(V_O^{\prime \prime}\right)\) will be created to compensate and achieve charge neutrality.
\[0.5 g \mathrm{Sc}_2 \mathrm{O}_3\left(\dfrac{1 \mathrm{molSc_{2 } \mathrm { O } _ { 3 }}}{137.9 g}\right)=0.0036 \mathrm{molSc}_2 \nonumber\]
\(0.0072 \mathrm{~mol} \mathrm{Sc} c^{3+}\) added \(=0.0036 \mathrm{~mol} V_O "\)
Total number of oxygen vacancies \(=2.18 \times 10^{21}\)
Lecture 24: Line Defects and Stress-Strain Curves
Summary
Line defects are 1-dimensional defects in a crystal that affect many macroscopic materials properties, including deformation. In 3.091, we’ll focus on two types of deformation: elastic deformation, which is reversible (strain only occurs when there is a stress applied, and it goes back once the stress is removed), and plastic deformation, which is permanent.

Elastic deformation can be likened to connecting all of the atoms with springs: Hooke’s law, F=-kx, tells us that there is a restoring force that returns the material to its initial state. This corresponds to the initial linear region in a stress-strain curve. If a material is brittle, it will likely break in the linear regime. However, if the material is ductile, it can undergo plastic deformation: the material no longer responds linearly to the applied stress. One deformation mechanism that occurs during plastic deformation is called slip: individual planes of atoms slide past each other due to the presence of a line defect. A dislocation is a type of line defect that forms when atoms are slightly misaligned: an extra plane of atoms exists in the crystal.

Dislocations can move through a crystal when a force is applied. Atoms slip over another to relieve the internal stress caused by applying a force. How can we tell what the slip planes are? In order for the material to slip, two adjacent planes of atoms must slide past each other. As this happens, the bonds between the planes must break and re-form. Therefore, slip planes must be planes that have the lowest inter-planar bond density. You can verify that this means that the most densely packed planes will be slip planes, because they have the highest intra-planar bond density. This means that slip occurs parallel to the closely packed planes: together, the slip planes and slip direction form a crystal’s slip system. Many materials can undergo dislocation-mediated slip, but metals in particular are known for this mechanism of deformation. The sea of electrons we learned about in metals allow bonds to move around (break and re-form in a new spot) with ease compared to ionic and covalent solids, which have much more rigid electronic structures.
When stress is first applied to an elastic material, dislocations form initially as planes of atoms pull apart. If two dislocations run into each other as they’re moving around, the defects can become pinned, so they can no longer move through the crystal. As more and more dislocations are pinned, they become tangled and slip doesn’t occur: the material would continue to deform elastically but it would require much more force to deform it. In other words, dislocation pinning makes materials harder. This mechanism of material strengthening is called work hardening. Work hardening causes the yield strength to increase, but at the expense of ductility.
Why this matters
Let’s talk about wind energy. Wind has the same intermittency challenges that solar has (so in other words it can only really be useful on a large scale if we can store the energy cheaply and efficiently). But wind also has many advantages and is a very appealing way to generate electricity. That’s why the installed wind capacity has seen tremendous growth globally over the last 20 years. In the U.S., more than 5% of our total electricity is now generated by the wind.
Global Cumulative Installed Wind Capacity 2001-2016 (pg. 3) chart
A wind turbine is actually based on fairly simple technology, which is one of the reasons it’s so appealing. Basically, the wind turns the blades, which provides the force to run an electric motor in reverse to generate electricity. The challenge is that the most consistent, high energy winds occur at high altitudes, so the blades are more efficient the larger and higher up one can make them go. This means that the blades have to support tremendous mechanical loads from the high wind power. And to make matters more difficult, the blades have to be able to go back and forth between high, low or no wind depending on the time of day. (as an aside, I love to look at maps and if you’re interested in seeing how the wind speed varies around the globe, there are a lot of cool maps that show this to you).
So in other words, we need blades that are strong enough that they don’t break apart under the extreme stress of the wind force, but flexible enough that they can bend without irreversible deformation. And what is it that we need to understand and engineer in order to make better blades? The line defects of course!!
Take a look at this plot of materials.

Here we’re looking at the density of the material on the x-axis and the Young’s modulus on the y-axis. The Young’s modulus is simply a measure of the elastic stiffness of the material, so if we go back up to our stress/strain curve at the beginning of this chapter, it’s related to the slope of the linear elastic regime. For a wind turbine blade, we’d like it to be lightweight and strong, but also flexible. As we learned in this lecture, the line defects are related to the plasticity, which is related to the yield stress, which is related to how much a material can be elastically strained.
From the chart, we can compare the different classes of materials (like metals vs. ceramics vs. polymers, etc) for two of these properties: density and stiffness. What’s interesting about this is that there are enormous parts of the chart that are currently empty. In other words, there’s a lot of work to be done to find and prepare materials that are both heavy and flexible, or light and strong. And you know what this means: new chemistry combined with control over line defects!
Why this employs

Let’s go big. This boat, for example, is called the SS Schenectady, and it has a bit of a problem. Mostly that it’s cracked in half. A lot of ships built during World War II were made from low-grade steel, which was too easy to fracture at the temperatures of the sea. In fact, they did test the strength of their steel but only in the dry dock, so the test temperature was higher than the operational temperature. Once the ship was in the colder water, the steel became much more brittle and it became much easier for defects to form, and once they formed it was easy for them to grow under applied stress. One certainly doesn’t want plastic deformation of a ship’s metal, but one does not want this type of brittleness either. Getting the right strength for the right application under the right conditions requires a lot of knowledge of the materials and their defect properties. And there are a lot of jobs in this space too. Think about it: pretty much everything we build today has to have some sort of operating conditions where the mechanical properties can be counted on to work as expected. Otherwise, ships crack in half, bridges collapse, and roads buckle.
Since I just gave the example of the ship, how about this one: the U.S. Department of the Navy has an opening for a “Materials Engineer” where they explicitly ask for experience in, “strength of materials (stress-strain relationships).” Corning’s Manufacturing, Technology and Engineering division is hiring a “Scientist/Engineer” to do modeling of the strength of materials, presumably mostly glasses. There’s an opening for a “Research Engineer – Materials Behavior,” at GE to design mechanical properties of new structural materials and coatings being for aircraft propulsion. There are so many jobs in this space I couldn’t possibly even categorize them all: pretty much any company that makes or deals with materials has jobs available related to their mechanical strength and failure. From ships, to buildings, medical devices to clothing, spacecraft to furniture, defects hold the key.
Extra practice
1. You obtain the following stress-strain curve for an aluminum sample (FCC).
a) Label the following regions on the plot (and the axes!):
Elastic regime Plastic regime Yield point Fracture point Elastic modulus

- Answer
-
Axes: stress, \(\sigma\) on the \(y\)-axis, has units of force/area. Strain, $\epsilon$ on the x-axis, is unitless, but often represented as length/length

b) What is the slip system in aluminum?
- Answer
-
The slip system for FCC is the close-packed direction and close-packed plane: \(\langle 110\rangle\) and \(\{111\}\) Recall that the angle brackets are used to denote families of directions, and the curly braces are used to denote families of planes!
Lecture 25: Amorphous Materials: Glassy Solids
Summary
Glasses are “amorphous materials:” all of the atoms are randomly arranged in a non-repeating structure. In \(3.091\), we'll focus on one type of glass: silica, or \(\mathrm{SiO}_2\). Each silicon atom has four valence electrons, so it is happy to form 4 single bonds. If an oxygen bonds to each of these valence electrons, each of the oxygens is left with an extra electron, forming a \(\left(\mathrm{SiO}_4\right)^{4-}\) molecule. However, when a solid is formed from these silicate molecules, the \(\mathrm{O}\) can be shared between neighboring silicates, forming a bridge.

The individual tetrahedral silicate molecules stay intact, but they can freely rotate relative to the other silicate molecules in the solid. When they don’t arrange in an ordered fashion, silica glass is formed. Whether the solid that forms is crystalline or glassy depends strongly on the processing conditions the silica undergoes.

One metric to quantify the processing conditions is by looking at how the molar volume changes as a function of temperature. For a crystal, the plot looks like this. There’s a sloped line that corresponds to the solid material, then a jump, then a different sloped line that corresponds to the liquid phase. The jump occurs at the melting temperature \(T_m\): when the material melts or freezes, it undergoes a huge change in volume. The slope of each line is defined as the coefficient of thermal expansion. However, sometimes when a material is cooled, it can remain in the liquid phase below \(T_m\): this is called supercooling. When a liquid is supercooled, continues to act like a liquid until one of two things happens:
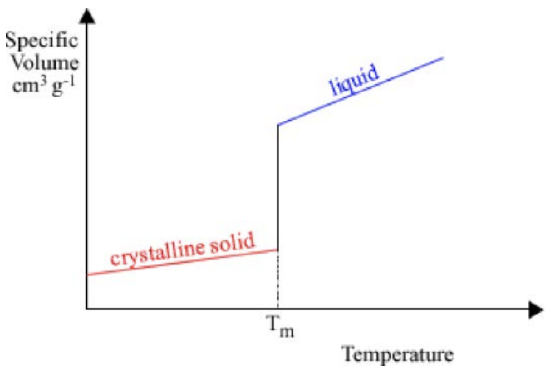
1. It crystallizes, characterized by a big jump down to the crystalline solid line and then solid behavior
2. It suddenly becomes a solid, “freezing” in its disordered state and becoming a glass. This transition is characterized by a change in slope at \(T_g\), the point at which solid forms (called the glass transition temperature), but no discontinuity in the freezing curve.
How can we know which path a material will take? It depends on materials properties: if the liquid has a high mobility (low viscosity), the molecules can move around easily and arrange into the energetically-preferential crystalline structure. Highly viscous or low mobility liquids are much more likely to get stuck in a glass. Further, if the crystalline structure is very complicated, or if the liquid is cooled very quickly, it’s hard for the atoms to find crystalline sites before the solid forms: these cases are also more likely to lead to glass formation.
The volume per mole is a good measure of the disorder in the material: the further the molar

volume is from the crystalline case, the more glassy the material is. Although a material only has one melting point, it can have multiple glass transitions depending on how it is processed. XRD is one tool that can be used to determine whether a material is a crystal or a glass: as the material gets more and more disordered, the sharp peaks observed in the XRD pattern disappear into a broad amorphous halo.
Why This Matters
\(\mathrm{SiO}_2\) glass is made from sand and there's a whole lot of it on the planet. \(\mathrm{SiO}_2\) stands out as a base material that has really awesome properties. That's partly because of its tremendous abundance. Check out this plot of the abundance of atoms in the earth's crust. Note that of all of the elements in the periodic table, oxygen and silicon are #1 and #2. This means that silica is cheap, and we're not going to run out, unlike other elements. In fact, there are many "critical elements" like \(\mathrm{Li}\) \(\mathrm{Co}\), \(\mathrm{Ga}, \mathrm{Te}\), and \(\mathrm{Nd}\) to name a few, labeled as such by the Department of Energy because there is concern that there will not be enough of them in the future to meet global demand. But \(\mathrm{Si}\) and \(\mathrm{O}\) are the opposite of critical: they are dramatically abundant. And this presents tremendous opportunity to use \(\mathrm{SiO}_2\) as a base material for wide-ranging applications.
And this is why the chemistry that we learned in this lecture matters: because we learned that

the key properties of glass, and the way that it’s processed, all come from the chemistry. The ways in which we continue to use glass on this planet can increase as human population and technological needs/use continue to increase, but how can we do utilize glass more sustainably given that the base material is so abundant? Or what other applications can glass be useful where it’s currently not used today? Can glass be made “greener”? One cool example for how this could work is in the work of Markus Keyser, who invented the “Solar Sinter.” This is a self-sufficient 3D printer of glass objects that you can drive out into the dessert, feed the sand and sunlight that are both plentiful, and print glass objects. The sunlight is used both to power the electric motor of the printer but also the create enough thermal energy to get above \(T_g\) for the sand. You can now buy commercial 3D glass printers, but I like this example because it’s emission-neutral.
Photo of Marcus Kayser's Solar Sinter
The future of how far we push technologies like this will depend on how far we’re able to push the properties of glass. In the next lecture we’re going to discuss a few different ways to control glass properties, but you only need today’s lecture and an understanding of those silicate groups to understand the key link between chemistry and why this matters.
Why this employs
Corning is one of the biggest and most well-known glass makers in the world. That video I mentioned in the lecture above that I showed in class, about Prince Rupert’s Drop, that was from the "Corning Museum of Glass" educational series. They are really, really into glass. And they're big: their 2018 revenue was \(\$ 11.4 \mathrm{~B}\) and they've got 51,500 employees currently. On their website if you click on the "engineering" section of the job openings page you'll find hundreds listings. They've also got a ton of internships for students:, "A Corning internship offers valuable hands-on experience for individuals in their chosen discipline to include but not limited to Material Science, Engineering, Research, Manufacturing, IT, HR, Marketing, Finance and Supply Chain." It's a cool place that has made it big out of glass and made glass into a big deal.
Corning works on a wide range of applications of glass, but let's focus on just one of them: fiber optic cables. Most of the backbone of today's internet is served by fiber optics, which are made of silica glass, because they have so many advantages over (older) copper wiring. For example, fiber optic cables can carry much higher bandwidth over longer distances than copper, this means the need for signal boosters is lessened, and fiber optic cables are also less susceptible to interference from external electromagnetic fields so they don't need shielding, and finally they don't break down or corrode nearly as often so they're much less expensive to maintain. It's no wonder that major U.S. companies like Comcast FiOS and Google Fiber are working to get fiber optic cables beyond being just the internet backbone, but into literally every single building in the country. Unlike Corning which many of us may have heard of already, some of the top fiber optics companies are less known even while being huge companies (meaning: lots of jobs!). Take OFS Optics, which makes fiber optics cables for over 50 different application spaces. They have over \(\$ 250 \mathrm{M}\) in annual revenues, and in one of their job postings for an "RD Engineer" states that they are looking for someone to, "lead the development of manufacturing processes for the next generation of glass optical fiber products." Cool.
Or how about going international to companies like Prysmian (based in Italy), which has over \(\$ 1 \mathrm{~B}\) in annual revenue, has a "Graduate Program" for recent grads to immerse them quickly with a mentorship program and also has the coolest name for a fiber with their "BendBright" brand. Or there's YOFC based in China with over \(\$1 \mathrm{~B}\)/year in revenue, and nice slogan, "Smart Link Better Life," or Fujikura with \(\$ 7 \mathrm{~B}\) annual sales and a claim to be, "Shaping the Future with Tsunagu Technology." (that means "connecting"). These and so many more companies are working on making next-generation fiber-optic cables, and if you dig a few layers deep into any of them, you'll see how complicated the production of fiber optic cables is, how many different ways it can be done today and will be done in the future, and how many jobs there are that directly relate to knowledge of \(\mathrm{SiO}_2\) glass!
It's not just all about processing: there is fundamental chemistry research to be done on silica glass, too. By doping silica glass with other elements, its properties can be changed. For example, by adding Erbium ions, the glass transforms from a passive light carrier to an amplifier capable of making the signal several orders of magnitude higher. And if you're interested in coding, there's a lot of work to be done to simulate how light travels in media like glass, and how it interacts with these dopants.
Extra practice
1. You obtain the following free volume vs temperature curves for a material cooled at three different rates. Label all instances of the following phenomena on the plot (and the axes!):
a) \(T_g\)
b) \(T_m\)
c) glassy regime
d) crystalline regime
e) fastest cooling rate
f) slowest cooling rate
g) liquid
h) supercooled liquid

- Answer
-

Lecture 26: Engineering Glass Properties
Summary
What does it mean to engineer glass? It can mean adding impurities that change properties like the glass transition temperature \(\left(\mathrm{T}_g\right)\), the solubility, the durability, etc. What unites most of these glass modifiers is that they are oxide donors, meaning they give up an \(\mathrm{O}^{2-}\) ion. This implies that these modifiers have stable cations, so often metals are good. For example:
\(\mathrm{CaO} \rightarrow \mathrm{Ca}^{2+}+\mathrm{O}^{2-} \quad \mathrm{Na}_2 \mathrm{O} \rightarrow 2 \mathrm{Na}^{+}+\mathrm{O}^{2-} \quad \mathrm{Al}_2 \mathrm{O}_3 \rightarrow 2 \mathrm{Al}^{3+}+3 \mathrm{O}^{2-}\)
The donated \(\mathrm{O}^{2-}\) ion attacks the \(\mathrm{Si} - \mathrm{O} - \mathrm{Si}\) bond and breaks it into two. It's like a knife that cuts the glass bond, and so this process is called chain scission. The \(\mathrm{O}^{2-}\) is able to insert itself into the bond and with its two extra electrons satisfies the charge state of the oxygen atoms that now "cap" the chains on each end. So we have that \(\mathrm{Si}-\mathrm{O}-\mathrm{Si}+\mathrm{O}^{2-} \rightarrow \mathrm{Si}-\mathrm{O} \mid \mathrm{O}-\mathrm{Si}\) with negative charge on each \(\mathrm{O}\). As shown in the figure, the \(\mathrm{Na}+\) ions hang around the oxygen. The effect of chain scission on the properties of glasses is enormous. Just take the melting temperature as an example: for crystalline \(\mathrm{SiO}_2\) (quartz) the \(\mathrm{Tm}\) is greater than \(1200^{\circ} \mathrm{C}\). For soda-lime glass, the glass transition temperature is typically around \(500^{\circ} \mathrm{C}\). If the silicate chains are cut, then the material is much less viscous, and it can find better packing more easily, leading to lower volume per mole and also a lower glass transition temperature (more supercooling).
The base chemistry of the solid, which in the case described above is \(\mathrm{SiO}_2\), is the network former. The oxide donor is called the network modifier. Adding network modifiers is another way to change a glass cooling curve. For example, curve (b) to the right could be obtained using \(\mathrm{SiO}_2\) with \(5 \% \mathrm{PbO}\) and curve (a) using \(\mathrm{SiO}_2\) with \(10 \% \mathrm{PbO}\). The reason is that more cutting of the chains makes the material less viscous, which means it can find better packing and be supercooled more.

We discussed two ways that mechanical properties are engineered in glass. First, the glass can be tempered: molten glass is cooled down with air and if the outside of the glass solidifies while the inside is still a liquid, then the outside has a completely different volume per mole than the inside. the hot melted \(\mathrm{SiO}_2\) solidifies but since it cannot have the smaller volume that it would like to have, it puts a inward pressure (compressive stress) on the already-solid outside layer. The second method of glass strengthening is called ion exchange. It involves swapping ions left in glass by network modifiers with ions of different size, which creates compressive stress.
Why this matters
The ability to engineer glass with wide-ranging properties has led to its use in a whole lot more than windows. How about: doors, façades, plates, cups, bowls, insulation, food storage, bottles, solar panels, wind turbines, mirrors, balustrades, tables, partitions, cook tops, ovens, televisions, computers, phones, aircraft, ships, windscreens, backlights, medical technology, optical glass, biotechnology, fiber optic cables, radiation barriers. And on top of that, glass is almost fully recyclable. The main reason glass has become so ubiquitous in all of these different ways is because of its massive chemical tunability as discussed in this lecture.

But here I want to focus on one particular property: strength. We talked today about using compressive stress to make glass stronger. But what if glass could be made stronger still? What if it could be made stronger than major structural materials like steel? In research labs, that is exactly what is happening. For example, in a Nature Materials paper from 2011 (doi:10.1038/nmat2930), the authors made a certain type of metallic glass stronger than steel and critically also tougher than steel. That means that not only does it have a high Young’s modulus, but when it breaks it can deform plastically as opposed to shattering. They created this new material through its chemistry, by adding a touch of palladium and a dash of silver to the mix. It already had a bit of phosphorus, silicon, and germanium, but by adding the palladium and silver, the glass was able to surpass steel in both hardness and toughness. Since then, many more demonstrations of mechanically super-strong glass have been made (often trying to avoid Palladium which costs $50,000 per kg. Here’s a plot from that same paper, showing the fracture toughness vs. the yield strength of different materials. Again, the yield is related to how much force the material can withstand without breaking, and the toughness is how much it can break without shattering. Going up on both axes can be very appealing for many applications. I love plots like this (called “Ashby plots”) since we can right away compare a bunch of different materials, in this case oxides, ceramics, polymers, metals, and of course their own new stuff (shown as “x” marks on the plot). Note how strong regular old oxide glasses are but also how little toughness they have (when they give, they shatter). But notice also how much tougher they can get by engineering their chemistry. This could put amorphous materials on a trajectory to becoming some of the most if not the most damage tolerant materials in the world!
Why this employs
In the last lecture for this section I listed glass manufacturing and companies working on innovating in glass chemistry. For this chapter on engineered glass, let’s talk about smarts. In particular, “smart glass.” For now, that label means one specific type of silicate-based glass: switchable glass. It has been around for a long time, as even in the 1980’s you may have noticed (ok, your parents may have noticed) people wearing the sunglasses that automatically tinted and de-tinted in response to the sun (that used what are called “thermochromic” materials embedded in the glass, which change color based on temperature. They never worked all that well, staying a little too shaded inside and a little too unshaded outside, but the idea was there. But now we’ve gone from thermo- to electro-chromic glass, and the possibilities are seriously exciting. With a tiny applied voltage, glass can be engineered to go back and forth between near full transparency and near full-opacity. Apart from being extremely cool, this type of technology can have a lot of positive sustainability-related benefits, since the glass can be programmed to automatically dim and brighten in response to outdoor light conditions — that can in turn dramatically reduce a building’s energy needs.
This type of smart glass is still on the early side although a number of companies are taking off, and that means jobs. These will be jobs at either mid or early-range start-ups, but in some cases they’ve closed mega (>$100M) fund-raising rounds so definitely growth is strong. Some companies in this space include Kinestral, Smartglass, View, Suntuitive, Gentex, Intelligent Glass, or Glass Apps. A lot of the investment in these companies is coming from the bigger ones like Asahi Glass or Corning, which have of course also started their own smart glass programs. Taken together all of this spells jobs in the future of glass. And its future looks very bright, far beyond switching the color or transparency, as the thermal, electronic and optical properties of the material continue to be engineered. We may or may not be living in the “Age of Glass,” as Corning likes to say, but we sure are living in an exciting time for this material.
Example Problems
1. The 2-D structure of soda-lime glass (used in windows) is shown below.
a) What compounds were used to make this glass? Do these compounds serve as network formers or network modifiers?
- Answer
-
\(\mathrm{SiO}_2\) : network former
\(\mathrm{CaO}\) : network modifier
\(\mathrm{Na}_2 \mathrm{O}\) : network modifier
b) How do each of the added compounds impact the bond structure in the glass?
- Answer
-
\(C a_O\) : breaks one bond/creates two network modifiers (coordinated with \(1 \mathrm{Ca}^{2+}\) ion)
\(\mathrm{Na}_2 \mathrm{O}\) : breaks one bond/ creates two network modifiers (coordinated with \(2 \mathrm{Na}^{+}\) ions)
3. If they are cooled at the same rate, would you expect silica glass with 14% \(\mathrm{Na}2\mathrm{O}\) or 25% \(\mathrm{Na}2\mathrm{O}\) to have a:
Higher molar volume?
Higher glass transition temperature?
Higher viscosity?

- Answer
-
\(14 \%\) would have the higher molar volume
\(14 \%\) would have the higher glass transition temperature
\(14 \%\) would have the higher viscosity
4. If a silica glass is doped with \(\mathrm{MgO}\), and then ion exchange is performed such that \(\mathrm{Ca}\) ions replace the \(\mathrm{Mg}\) ions, how would the mechanical properties of the glass change?
- Answer
-
\(\mathrm{Ca}\) ions take up more space than the \(\mathrm{Mg}\) ions, so the glass will be under internal compression (like the Prince Rupert's drop)
Lecture 27: Reaction Rates
Summary
Chemical kinetics means the study of reaction rates, which correspond to changes in concentrations of reactants and products with time. Some terms to know: concentration \(=\) moles / liter \(=\) molarity \(=[]\), rate \(=\mathrm{d}[] / \mathrm{dt}\), a rate law is some equation that relates the rate to [], an integrated rate law relates the [ ] to \(\mathrm{t}\) (ime), and the Arrhenius equation gives us the rate vs. \(\mathrm{T}\)(emperature)
Take a simple reaction where \(a A \rightarrow b B:\) since mass is conserved, A disappears no faster than \(\mathrm{B}\) appears, so the actual reaction rate is \(=1 / \mathrm{b} \mathrm{d}[\mathrm{B}] / \mathrm{dt}=-1 / \mathrm{a} \mathrm{d}[\mathrm{A}] / \mathrm{dt}\). In other words, the change in the concentration of \(\mathrm{B}\) must equal the opposite of the change in concentration of \(\mathrm{A}\) weighted by one over the molar coefficient a or b. We can have more than one reactant product and the same idea holds. For example, suppose we have 2 of each: \(a \mathrm{~A}+\mathrm{bB} \rightarrow \mathrm{cC}+\mathrm{dD}\). In this case the reaction rate would be:
\(\text { rate }=\dfrac{-1}{a} \dfrac{d[A]}{d t}=\dfrac{-1}{b} \dfrac{d[B]}{d t}=\dfrac{1}{c} \dfrac{d[C]}{d t}=\dfrac{1}{d} \dfrac{d[D]}{d t}\)
The general way to write an equation for the rate for the equation above is: rate \(=\mathrm{k}[\mathrm{A}]^m[\mathrm{~B}]^n\), where \(\mathrm{k}=\) rate constant and is dependent on conditions (\(\mathrm{T}, \mathrm{P}\), solvent), m and n are exponents determined experimentally, \(m+n\) is called the reaction order. Note that the rate units must always be M/s by definition, so this means that units of \(\mathrm{k}\) depend on \(\mathrm{n}\) and \(\mathrm{m}\). For this class we’ll cover three different orders of reactions: \(0^{th}, 1^{st},\) and \(2^{nd}\).

To know the order of a reaction based on data tables like the one below, take any two rows of data: say the \(\mathrm{t}=26 \mathrm{~min}\) and \(\mathrm{t}=70 \mathrm{~min}\) rows. The concentration ratio between these two times is \(0.0020 / 0.0034=0.5882\). The rate ratio is \(1.8 / 5.0=0.36\)

First of all the rate is changing so it can’t be 0th order. Second of all, at two different times the ratio of concentrations is not equal to the ratio of rates, so it can’t be 1st order. But if we square the ratio of concentrations, \((0.0020/0.0034)^2 = 0.35\) which is very close to \(0.36\), so now we have our answer: from the data we can say the reaction is 2nd order!
To know the role of temperature in determining reaction rates, we must first learn about collision theory. Collision theory frames the reaction between molecules, say A and B, as follows: 1) a reaction can only occur when A and B collide, 2) not all collisions result in the formation of product, 3) there are two factors that matter most: the energy of the collision, and the orientation of molecule A with respect to B at the time of collision.

We can think of the energy required for \(\mathrm{A}\) to react with \(\mathrm{B}\) to be a kind of “activation energy” or \(\mathrm{E}_a\). As we learned in chapter 14 (phases), molecules at a given temperature have a distribution of kinetic energies with that temperature being the average. That means some molecules have much more energy than the average, while other have less. Reactions are similar in that it’s the part of the distribution higher than the activation energy that matters. This plot shows how this works: the distribution of energies for a given molecule at two different temperatures shows that for higher temperature more molecules will have energies above the activation energy than for the lower temperature.
The Arrhenius equation gives us an expression that summarizes the collision model of chemical kinetics. It goes as follows: rate = (collision frequency)*(a steric factor)*(the fraction of collisions with \(\mathrm{E} > \mathrm{E}_a\)). In math terms, that’s shown here for the equation for rate (\(\mathrm{k}\)).

\(\mathrm{A}=\) the frequency factor, and its units depend on the reaction order. For example if the reaction is first order then the frequency factor must have units of \(s^{-1}\) The activation energy, \(\mathrm{E}_a\), we've already discussed. If it's given in units per mole, like \(\mathrm{J} / \mathrm{mol}\), we use \(\mathrm{R}\) as it's written, where \(\mathrm{R}\) is the ideal gas constant \(\mathrm{R}=8.314 \mathrm{~J} / \mathrm{K}^*\) mol. If the activation energy is given in units of \(\mathrm{eV}\), then the constant used would be the Boltzmann constant in units of \(\mathrm{eV}\left(8.61733 \times 10^{-5} \mathrm{eV} / \mathrm{K}\right)\).
This relationship means that if we plot the natural \(\log\) of the rate vs. \(1 / \mathrm{T}\) then it should be a straight line with slope \(=-\mathrm{Ea} / \mathrm{R}\) and intercept \(=\ln (\mathrm{A})\), as shown in the plot above.
So we’ve covered concentration, and now temperature. The last example of a way to change the rate of a reaction that we’ll mention (and unlike those other two, we’ll really just mention it and not go into detail) is the catalyst. A catalyst is a way to increase the rate of a reaction without having anything consumed as part of it. It’s a material that, in the language of our discussion on Arrhenius above, lowers the activation energy for the reaction.
Why this matters
Let's keep going with the catalyst theme for this section. It is estimated that \(\approx 90 \%\) of all commercially produced chemical products involve catalysts at some stage in the process of their manufacture! Some of these processes I've already highlighted in other Why This Matters moments, like the Haber-Bosch process for fixing \(\mathrm{N}_2\), or the depletion of \(\mathrm{O}_3\) by CFCs. At the time, we hadn't learned about reaction rates or catalysts, so I didn't go into it. But in both cases the role of the catalyst is absolutely essential (in fact, the big innovation of Haber-Bosch was not to discover the reaction (which had been known) but rather to discover a catalyst that lowers the temperature needed to make the reaction happen economically and at large scales.
Let's discuss another world-changing catalytically enhanced reaction: namely, the removal of most toxic emissions from cars and trucks. I know, you may be thinking that the tailpipe of a car smells pretty toxic. And that's because it is, but it's a whole lot better than it used to be, and the reason is the catalysts that are now part of every tailpipe in the form of what is called the catalytic converter. Why did we need these in the first place? It all goes back to the very first reaction we wrote on the first day of lecture: combustion. One example I gave was the combustion of methane:
\(\mathrm{CH}_4+2 \mathrm{O}_2 \rightarrow 2 \mathrm{H}_2 \mathrm{O}+\mathrm{CO}_2\)
It's true that \(\mathrm{CO}_2\) is harmful to the environment for reasons of climate change, but there's nothing toxic in those products... so what's the problem?

Ah, if only cars burned pure methane! But gasoline is far, far away from a pure fuel source. And furthermore, even modern car engines are far, far away from being able to burn the fuel perfectly without side-reactions. Gasoline is a mixture of about 150 different chemicals, and these include not just those hydrocarbons that combust, but also a host of additives that range in purpose from corrosion inhibitors to lubricants to oxygen boosters. Since this complex chemical soup doesn’t burn cleanly, we get both direct products and by-products that go far beyond the pure case of \(\mathrm{H}_2\mathrm{O}\) and \(\mathrm{CO}_2\). Many of these products are pollutants and some are really bad ones. An incomplete list would include: carbon monoxide (\(\mathrm{CO}\)) which is poisonous, nitrogen oxides (like \(\mathrm{NO}\) and \(\mathrm{NO}_2\), or “\(\mathrm{NOX}\)” as they’re called) which cause smog and have many adverse health effects, sulfur oxides (yes, you guessed it, “\(\mathrm{SOX}\)”) which cause acid rain, and unburned hydrocarbons or volatile organic compounds (VOCs) which cause cancer.
Similar to the removal of CFC's from refrigerants, cleaning up the tailpipe represents a fantastic example for how policy and regulation can make the world better. It was the Clean Air Act that Congress passed in 1970 that gave the newly-formed EPA the legal authority to regulate this toxic mess that came out of cars. As a result, today's cars are \(\approx 99 \%\) fewer emissions compared to the 1960s! The fuels are also cleaner (lead was removed and sulfur levels lowered), and taken together cities have much healthier air. Take a look at this picture of New York city from 1973 (left) compared to 2013 (right). In the 1970's, smog from car exhaust overwhelmed most major U.S. cities.
The key technology that enabled this dramatic clean-up is the catalytic converter. Inside a catalytic converter there are actually multiple catalysts, each enhancing different reactions. Most cars today use what is called a "three-way" catalyst, which just means that it tackles all three of the biggest pollutants: NOX, hydrocarbons, and \(\mathrm{CO}\). The key materials used as the catalysts are a combination of platinum, palladium, and rhodium. Inside a catalytic convertor one typically has a honeycomb mesh just to get a large surface area. Since the temperature gets quite high (and in fact needs to be high for the catalysts to operate, which is why cold engines pollute more than hot ones), the honeycomb mesh is made out of a ceramic material like alumina so it can handle \(\mathrm{T}=500^{\circ} \mathrm{C}\) without cracking or degradation. The \(\mathrm{Pt}\), \(\mathrm{Pd}\), and \(\mathrm{Rh}\) metals coat the \(\mathrm{Al}_2 \mathrm{O}_3\) mesh, and the exhaust flows through.

Take a look at this catalytic converter schematic. You can see two different chambers, one where the metal acts as a "reduction catalyst" for the NOX removal and the other where a different metal (or combination of metals) acts as an "oxidation catalyst" to treat \(\mathrm{CO}\) and unburned hydrocarbons. In the reduction catalyst chamber, the reaction we're trying to accelerate is \(2 \mathrm{NO} \rightarrow \mathrm{N}_2+\mathrm{O}_2\). In order to do so, the catalyst binds the \(\mathrm{NO}\) molecule to it, and is then able to pull off the nitrogen atom from \(\mathrm{NO}\) and hold it in place. Then another \(\mathrm{N}\) atom that also got pulled off of a different \(\mathrm{NO}\) molecule gets stuck to the catalyst somewhere nearby, and those Likewise, the oxygen can form \(\mathrm{O})2\). The whole point is that the catalyst finds a different way to carry out the same reaction with much lower barriers. The oxidation catalyst burns \(\mathrm{CO}\) and hydrocarbons using remaining \(\mathrm{O} 2\) gas, for example to get this reaction to go: \(2 \mathrm{CO}+\mathrm{O}_2 \rightarrow 2 \mathrm{CO}_2\). Again, that reaction would not occur at a high rate normally, but the catalyst breaks it down into steps (like splitting \(\mathrm{CO}\)) that occur much more easily.
Why this employs
The rate of a reaction is of course of central importance to anything one does that involves a reaction. And how many different types of jobs do anything involving a reaction? A ton! We could be talking about any sort of chemical synthesis job, or in drug design where reaction rate is crucial to the drug’s effectiveness and safety, or how about food making (even beyond beer) where rates determine anything from how long to let dough rise to how fast that dough browns in an oven vs. the apples inside turning mushy (I just made an apple pie in case you couldn’t tell), to how quickly to food goes bad. The point is that making stuff is inherently about rates.
And so instead of focusing on any one of these things that we make, I’d like to use this Why This Employs section to mention the fact that in nearly everything we make today, far too much natural capital is expended, and it doesn’t have to be that way. A single computer chip takes more than 600 times as much mass to make it and tremendous amounts of both water and energy (including annealing multiple times at temperatures over 1000°C. Cement is a wonderful material (“liquid stone”) but it takes baking calcium silicate at very high temperatures to make the tiny particles called “clinkers” that give it the ability to take on any shape in an instant and then dry and set so quickly after. Why is it that, time and time again, we fail to do what nature does so well? Animals can build incredibly strong and complex structures without going a single degree over room temperature. Spiders spin silk stronger than steel without lighting a fire. How can we do better? Much of it comes down to reaction rates. We use these massive amounts of energy and high temperatures and harsh chemicals because we want to make stuff quickly and because we need to make stuff in almost unimaginable quantities. China alone produced 2.5 trillion kilograms of cement last year!
There are more sustainable ways to make stuff, especially if we can get a handle on the reaction rates. Ten years ago I remember reading articles about the new revolution of “green chemistry” (check out for example this 2010 article in Scientific American, https://www.scientificamerican. com/article/green-chemistry-benign-by-design/. I remember thinking then that this field was ready to take off. Well, it didn’t really happen with a bang, but slow and steady this idea of benign design is taking hold. If you search for green chemistry jobs, you’ll find many companies now investing in this area substantially and that spells new jobs. The idea of green chemistry on the job market ranges quite broadly, from finding ways to synthesize materials without toxic chemicals, to making the drug discovery process biodegradable, to lowering the temperatures needed in a manufacturing step. Whatever it is, it’s all about the rates.
Example problems
1. Acetic acid is made from carbon monoxide and methanol according to the following equation:
\(CO(g)+MeOH(g) \rightarrow A c O H(g)\)
Your company wants to know how to improve this reaction: they present you, a chemical consultant, with the following data. Propose a rate law for the reaction.
| \(\mathrm{CO}\) pressure | \(\mathrm{MeOH}\) pressure | Acetic acid formed |
|---|---|---|
| 21.5 atm | 15.0 atm | 980 mol/hour |
| 11.1 atm | 15.0 atm | 499 mol/hour |
| 11.1 atm | 10.0 atm | 502 mol/hour |
- Answer
-
The rate is fairly insensitive to changing \(\mathrm{MeOH}\), but the rate drops by about half when the pressure of \(\mathrm{CO}\) is halved. Therefore, the rate is \(0^{th}\) order in methanol, but \(1^{st}\) order in \(\mathrm{CO}\):
\(rate = k[CO]\)
Lecture 28: Equilibium and Solubility
Summary
Many of you have probably heard the expression, “like dissolves like”. . . but let’s add a little bit more chemistry to that phrase. What is really meant by the word “like” is “similar bonds,” so the expression could also be said, “similar bonds dissolve similar bonds.” Solubility is a metric that tells us how much the solute (the solid being dissolved) can break apart and dissolve into the solvent (the liquid in which the solid is dissolving). When the solubility is miscible that means that it can be full mixed together at any concentration.
Intermolecular forces play a huge role in solubility. Water is called a polar solvent (also called hydrophilic) because it’s a solvent with large dipole moments, while hydrocarbons are examples of nonpolar solvents (also called hydrophobic) since the dominant IMF is London. Polar solvents are in general immiscible with nonpolar solvents.
What’s really going on at the scale of the molecule and its various IMFs, is that if a molecule can lower or keep similar its energy by welcoming a different molecule into its bonding environment, then it will do so. If on the other hand the other molecule cannot bond in the same way (as in the example of the longer hydrocarbon chaining having much more London forces than H-bond ability) then it would raise the energy of the system for the solvent to bond to it over itself.
Particles in a solution that re-join the solid are said to be precipitating. both dissolution and precipitation occur simultaneously and at the point of saturation, or maximum solubility, they happen at exactly the same rate. This is called dynamic equilibrium.
We know from our previous lectures on ionic solids that these are cations and anions in the solid, which is the basis of the ionic bond. When dissolved in water, they also remain ions, just now in solution, where they interact strongly with water. We can understand why, since we know from our IMF lecture that the ions will interact with the dipole of the water molecule. The reason there is a trend in the solubility of these salts is related to the strength of the bond between ions in the solid relative to those the liquid. As the ionic bonding becomes stronger (from weakest at \(\mathrm{NaI}\) to strongest at \(\mathrm{NaF}\)), less of it dissolves.
Suppose the general reaction has already reached its dynamic equilibrium:
\(a A+b B \leftrightarrow c C+d D\)
This means that the four different concentrations, \([\mathrm{A}],[\mathrm{B}],[\mathrm{C}]\), and \([\mathrm{D}]\) are not changing even though the reaction is still going in both directions. Below is a nice picture of what might happen to these concentrations as a function of time. At first during the reaction the concentrations change, and then they all flatten out. At that point, when they're all flat, the system has reached dynamic equilibrium. We define the reaction quotient, \(\mathrm{Q}\), as the ratio of
\(\dfrac{[C]^c[D]^d}{[A]^a[B]^b}\)

This expression for \(\mathrm{Q}\) can be evaluated at any point during the reaction, whether the system has reached equilibrium or not. \(\mathrm{BUT}\), we have a special way to call \(\mathrm{Q}\) once the system is in dynamic equilibrium, and that's with the letter \(\mathrm{K}\), or the equilibrium constant for the reaction. Often, we put a subscript "eq" on the \(K\) just to make sure we know what it refers to, so \(\mathbf{K}_{e q}=\mathbf{Q}\) (for the reaction quotient in equilibrium). The Keq for a solid dissolving in water is the solubility product \(\left(\mathbf{K}_{s p}\right)\)

By comparing the value of \(\mathrm{Q}\) to the equilibrium constant, \(\mathrm{Keq}\), for the reaction, we can determine whether the forward reaction or reverse reaction will be favored.
If \(\mathrm{Q}=\mathrm{K}_{e q}\), the reaction is at equilibrium. If \(\mathrm{Q}<\mathrm{K}_{e q}\), like at point \(\mathrm{D}\), then the reaction will move to the right (in the forward direction) in order to reach equilibrium. If \(\mathrm{Q}>\mathrm{K}_{\text {eq }}\), like in point \(\mathrm{E}\), then the reaction will move to the left (in the reverse direction) in order to reach equilibrium. Le Chatelier coming at us in graphical form!
Why this matters
From examining the tiny bubbles trapped in ice cores, we know that the level of \(\mathrm{CO}_2\) currently in the atmosphere is more than it has been in over a million years, and the amount is growing exponentially. But while this incredibly important greenhouse gas has received tremendous attention for its role in climate change, there is a much less frequently discussed impact that \(\mathrm{CO}_2\) is having on our planet. Maybe that’s because it’s happening in the oceans, which we don’t live in so it’s harder to make as big of a deal about the changes occurring. But we will feel these changes soon enough. The ocean has absorbed more than 500 billion tons of \(\mathrm{CO}_2\) from the atmosphere, as it quietly captures roughly a quarter of what’s being emitted day in and day out (today we humans crank out 1,200,000 grams of \(\mathrm{CO}_2\) per second). So why does this matter?

The problem with all that extra \(\mathrm{CO}_2\) in the oceans is that it makes the water more acidic. Over the past 200 years alone, ocean water has become 30 percent more acidic. This change in acidity is faster than any known change in ocean chemistry over the last 50 million years! In geological terms, this is an extremely abrupt perturbation, and without dramatic changes in human behavior we are only seeing the beginning.
Unfortunately, one of the most important defenses for ocean creatures - the shell - cannot survive increased acidity. Here's a picture of what happens to the shell of a pteropod, a type of mollusk only a few \(\mathrm{cm}\) long, after 45 days in a solution of \(\mathrm{pH}=7.8\). Now, the oceans aren't quite that acidic yet (current ocean \(\mathrm{pH}\) is 8.1), but they're well on their way. Pteropods are one of only a few basic types of sea creature that sits at the very bottom of the ocean food chain, right above plankton and seaweed, which means that if their numbers are reduced, everything higher on the food chain is impacted, from krill to salmon, to herring and birds, to seals and polar bears, and of course, to us.
In the goodie bag that goes with these lectures, you’ll be able to see this dissolution in action. We make it go faster than 45 days since I don’t want you to wait that long, but the chemistry at play is the same. I wanted you to touch and feel this precious calcium carbonate loss, so that you can get a glimpse into what is beginning to happen to 2/3 of our planet.
Why this employs
In order for drugs to have a physiological effect on the human body, they must be in solution. In particular, they must be dissolved in solution if they start out in solid form, like tablets do. Now, the rate of the dissolution is extremely important since determines how fast the drug is absorbed in the body. Rates were discussed in the last chapter. But solubility itself (i.e. regardless of how fast it dissolves, what is its ability to fully dissolve at all?) is also of crucial importance in drug design. If the drug is not very soluble, formulations can lead to difficulties in the use of the drug which may otherwise have very beneficial effects. If a drug has low solubility that means it will likely have a low level of what is called “bioavailability,” which basically just means that it won’t get enough exposure in the body.
In pharmaceutical companies that do most of the new drug development, it is estimated that a whopping 40% of all new chemicals (possible new beneficial drugs) are practically insoluble in water, making them effectively useless. And what is it that these companies do to try to increase solubility, especially for otherwise promising candidates? Chemical modification, of course! And so it is that all of the large pharmaceutical companies are looking for people who know about solubility! In terms of where to look for jobs, the thing about drug design companies is that you don’t have to look very far. Within just 1 mile of campus, there are dozens of both large and small companies specializing in drug design. More broadly, Kendall Square now has the largest biotechnology industry per square foot, with over 120 companies within a mile.
In terms of which ones of these companies have jobs related to solubility, well because of how important solubility is, it’s pretty much all of them. Check out for example, the job postings at Takeda, Novartis, Alnylam, Sanofi, Pfizer, and Genentech just to get started, and then walk down the street to go learn more about the companies, what they do, and meet with them in person!
Example Problems
1. Calcium fluoride has a \(\mathrm{K}_{sp}\) of \(5.3×10^{-9}\). How much calcium fluoride can dissolve in 1\(\mathrm{L}\) of water?
- Answer
-
\[k_{s p}=[C a][F]^2 \nonumber\]
For every mole of \(\mathrm{Ca}\) that dissolves, twice as many moles of \(\mathrm{F}\) must dissolve since the chemical formula is \(\mathrm{CaF}_2\). Assuming \(\mathrm{x}\) moles of \(\mathrm{Ca}\) dissolve:
\begin{gathered}
K_{s p}=[x][2 x]^2 \\
5.3 \times 1-^{-9}=4 x^3 \\
x=0.00109 M \\
78 \dfrac{g}{\mathrm{~mol}} \dfrac{0.00109 \mathrm{~mol}}{L}=85.9 \mathrm{mg} / 1 \mathrm{~L}
\end{gathered}
Lecture 29: Common Ions and Acids/Bases
Summary

Le Chatelier’s principle states that the position of equilibrium moves to account for changes to the system, such as the introduction of a new ion to a solution. But what happens if we add a new compound that has one ion in common with an existing solution? The equilibrium curve for the \(\mathrm{AgCl}\) dissociation is shown to the right. Suppose we add \(\mathrm{NaCl}\): it will dissociate completely into ions \(\mathrm{Na}^{+}(\mathrm{aq})\) and \(\mathrm{Cl}^{-}(\mathrm{aq})\). We can use an ICE table to determine the shift in equilibrium: if we start at point \(\mathrm{B}\) and add \(0.1 \mathrm{M} \mathrm{NaCl}\), we can use the equilibrium constant for \(\mathrm{AgCl}, \mathrm{K}_{s p}=1.7 \times 10^{-10}\). As \(\left[\mathrm{Cl}^{-}\right]\) increases, \(\left[\mathrm{Ag}^{+}\right]\) must decrease to keep \(\mathrm{K}_{s p}\) constant.
| [\(AgCl\)] | [\(Ag^+\)] | [\(Cl^-\)] | |
|---|---|---|---|
| I | (all solid) | \(1.3 \times 10^{-5}\) | \(1.3 \times 1^{-5}\) |
| C | + x precipitates | \(-x\) | \(+0.1 - x\) |
| E | More solid | \(1.3 \times 10^{-5} - x\) | \(+0.1 - x\) |
\begin{gathered}
K_{s p}=\left[\mathrm{Ag}^{+}\right]\left[\mathrm{Cl}^{-}\right]=(0.1-x)\left(1.3 \times 10^{-5}-x\right) \\
K_{s p}=0.1\left(1.3 \times 10^{-5}-x\right)=1.7 \times 10^{-10} \quad x=1.7 \times 10^{-9}
\end{gathered}
This is called the common ion effect: the solubility of one constituent is repressed by the addition of a second solute.
For a generic case of any acid, call it "A" we write its dissolution reaction as follows: \(\mathrm{HA}+\mathrm{H}_2 \mathrm{O} \rightarrow\) \(\mathrm{H}^{+}+\mathrm{A}^{-}+\mathrm{H}_2 \mathrm{O}\). Although we write \(\mathrm{H}^{+}\) to make the proton dissociation explicit, we never actually mean that if it's in water. That's because the \(\mathrm{H}^{+}\)ion is not stable in \(\mathrm{H}_2 \mathrm{O}\), instead it always becomes \(\mathrm{H}_3 \mathrm{O}^{+}\). The first person to define acids and bases in any sort of more concrete chemical way was good old Svante Arrhenius. According to him, an acid is a substance that dissolves in water to produce \(\mathrm{H}+\) ions, and a base is a substance that dissolves in water to produce hydroxide \(\left(\mathrm{OH}^{-}\right)\)ions.
Arrhenius defined acids and bases in terms of the presence of \(\mathrm{H}^{+}\) or \(\mathrm{OH}^{-}\)ions in solution: for example, \(\mathrm{HCl}(a q) \rightarrow \mathrm{H}^{+}(a q)+\mathrm{Cl}^{-}(a q)\) is an "Arrhenius acid" while \(\mathrm{NaOH}(s) \rightarrow \mathrm{Na}^{+}(a q)+ O H^{-}(a q)\) an "Arrhenius base."
Sorenson came up with the "power of hydrogen" scale, or "pH" scale for short. \(\mathrm{pH}=-\log \left[\mathrm{H}^{+}\right]\). The \(\mathrm{pH}\) scale runs from 0 to 14 , with 0 being extremely acidic and 14 being extremely basic. We can do the same thing for the concentration of \(\left[\mathrm{OH}^{-}\right]\): the power of \(\mathrm{OH}^{-}\) in solution, or the "pOH" \(=-\log \left[\mathrm{OH}^{-}\right]\), with the opposite definition.
A strong acid like \(\mathrm{HCl}\) fully dissociates: \(\mathrm{HCl}+\mathrm{H}_2 \mathrm{O} \rightarrow \mathrm{H}^{+}+\mathrm{Cl}^{-}+\mathrm{H}_2 \mathrm{O}\), so \(0.1 \mathrm{M} \mathrm{HCl}\) has \(\left[\mathrm{H}^{+}\right]=0.1\), so \(0.1 \mathrm{M} \mathrm{HCl}\) solution has \(\mathrm{pH}=-\log [0.1]=1.0\).
Some materials can be both acids and bases. None other than water itself is one such material! When a molecule can either give \([\mathrm{H}+]\) ions or \([\mathrm{OH}-]\) ions into solution, it's called amphoteric: \(\mathrm{H}_2 \mathrm{O}(l)+\mathrm{H}_2 \mathrm{O}(l) \leftrightarrow \mathrm{H}_3 \mathrm{O}^{+}(a q)+\mathrm{OH}^{-}(a q) . \mathrm{K}_{s p}\) for water is called \(\mathrm{K}_w: K_w=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{OH}^{-}\right]= 1.0 \times 10^{-14}\). If an acid is added to pure water the hydrogen ion concentration increases (and the \(\mathrm{OH}\) ion concentration decreases) A base goes the other way, adding hydroxide ions into the pure water solution. All of the tricks we learned for \(\mathrm{K}_{s p}\) apply to \(\mathrm{K}_w\), which is especially useful in the context of acids and bases.
Arrhenius' definition of acids and bases has two key limitations: first, because acids and bases were defined in terms of ions obtained from water, the Arrhenius definition applied only to molecules in aqueous solution. Second, and more important, the Arrhenius definition predicted that only materials that dissolve in water to give \(\mathrm{H}^{+}\) and \(\mathrm{OH}^{-}\)ions can have the properties of acids and bases. But there are many examples where this is not true! We need to go beyond Arrhenius to understand some acids and bases.
Why this matters
Now that we're armed with the concepts of the Common Ion Effect and Le Chatelier's principle, we can go into more detail on the chemistry that's causing the possible catastrophe discussed in the previous Why This Matters: ocean acidification and its impact on the fate of calcium carbonate. It's all about equilibrium and how \(\mathrm{CO}_2\) is shifting it in the oceans. The first reaction we consider is what happens when the oceans encounter more \(\mathrm{CO}_2\) from the atmosphere: namely, the \(\mathrm{CO}_2\) dissolves in the \(\mathrm{H}_2 \mathrm{O}\) to form carbonic acid:
\(\mathrm{CO}_2+\mathrm{H}_2 \mathrm{O} \leftrightarrow \mathrm{H}_2 \mathrm{CO}_3\)
So more \(\mathrm{CO}_2\) getting dissolved in the ocean means there's more carbonic acid in the ocean, which then produces dissociated ions as follows:
\(\mathrm{H}_2 \mathrm{CO}_3 \leftrightarrow \mathrm{HCO}_3^{-}+\mathrm{H}^{+}\)
But on the other hand, we've got solid \(\mathrm{CaCO}_3\) which is the calcium carbonate, that is in a nice equilibrium with the following dissociation reaction:
\(\mathrm{CaCO}_3 \leftrightarrow \mathrm{Ca}^{2+}+\mathrm{CO}_3^{2-}\)
The equilibrium constant for the dissolution of \(\mathrm{CaCO}_3\) is \(\mathrm{k}_{s p}=5 \times 10^{-9}\). That means not a whole lot of it will dissociate in "normal" (i.e. last \(>50\) million years) ocean conditions. The reason this equilibrium is getting thrown off is that there are now extra \(\mathrm{H}^{+}\)ions that come from the \(\mathrm{CO}_2\) reaction above. These \(\mathrm{H}^{+}\)ions consume (react with) \(\mathrm{CO}_3^{2-}\) to form \(\mathrm{HCO}^{3-}\) which lowers the concentration of \(\mathrm{CO}_3^{2-}\). Ah, but we just learned about that with the Common Ion Effect: if we add or take away some molecule that's part of a balanced equilibrium, then we will drive that equilibrium to counter whatever we've done. In this case , \(\mathrm{CO}_3^{2-}\) is getting consumed more than before, which in turn drives more dissolution of the \(\mathrm{CaCO}_3\). That's why the shells are dissolving, and it's why I want you to experience this chemistry directly in your goodie bag. What you're seeing is those experiments is an accelerated version of what is happening in the oceans.
Acidity is measured by " \(\mathrm{pH}\)," which is a logarithmic scale. Since the Industrial Revolution, the \(\mathrm{pH}\) of the oceans has decreased by \(\approx 0.1\) to \(8.07\), which is equivalent to a \(\approx 30 \%\) increase in the oceans' acidity. I showed that trend in last lecture's Why This Matters. Estimates are that at our current rate of \(\mathrm{CO}_2\) emissions, the acidity of the oceans will reach a whoppingly more acidic state of \(\mathrm{pH}=7.7\) by 2100 . That would represent a \(>5\)-fold increase in acidity compared to pre-industrial levels, and that would also pretty much do it in terms of wiping out most ocean life.
The ocean has absorbed roughly 525 billion tons of \(\mathrm{CO}_2\) from the atmosphere over the past 200 years, at a pace of \(\approx 22\) million tons per day. The change that this induces is the fastest known chemical change the ocean has experienced over the past 50 million years and probably much longer. Around 250 million years ago, at the end of what is known as the "Permian era," records show that there was a sudden change in the oceans, and geologists refer to what happened as, "The Great Dying." This is because more than \(90 \%\) of all marine life disappeared from the fossil record. Most scientists who study this kind of thing relate the mass extinction to a huge spike in volcanic activity, which put vast clouds of acidic dust into the atmosphere, which then fell into the oceans and raised their acidity.
The reason the dissolution of calcium carbonate is so important is that it is essential not just to the pteropods I showed before, but to so many other “base” elements of the ocean’s food chain, like phytoplankton, shell fish (mussels, oysters, lobsters, crayfish, etc), and of course coral, to name only a few examples. The calcium carbonate structures in these sea creatures are highly sensitive to slight changes in acidity, and without them the health of the oceans will fail. I’ll end this Why This Matters with a thought-provoking quote from Ken Caldeira who is a climate scientist at Stanford. In trying to help people get perspective on what we’re doing, he said, “Well, if the Romans had industrialized, it now would be two thousand years later. The seas would still be rising and still be completely acidified, and, yes, maybe they would have got a century or so of higher G.D.P. than they would have otherwise.” The point being: the deleterious changes we’re on track to make in the oceans won’t just impact our kids, and our kids’ kids; it will impact tens and more likely hundreds of generations to come.
Why this employs
I talked about ocean acidification in this lecture since we’re introducing acids and bases, and what’s going on in the oceans is a good example of the effects of chemistry happening on a planetary scale. But what if you want to actually work on this problem, like as a job? Well, that’s not as straightforward as you might think, or for that matter may have hoped, but there are some things you can do.
For one thing, there are many, many academic or state-run programs that are studying ocean chemistry, and many of them have postings for jobs. This ranges from a random ocean acidification project at the University Di’ Bologna, Italy, to the National Oceanic and Atmospheric Administration Cooperative Science Center for Earth System Sciences and Remote Sensing Technologies (yes, thankfully they go by NOAA-CESSRST), to other government agencies such as the EPA or any agency working in conservation and policy, to Washington State’s Department of Ecology, to Stony Brook University who is currently seeking an, “Ocean Acidification Monitoring Associate.” There are many faculty positions in this area, and I believe there will be more in the future. So if you’re interested in both this topic as well as a career in academia, then the path of research in scientific centers is a good one. There are a number of different specializations that would lead to studying (and hopefully helping to save!) the oceans, from marine biologist to environmental engineer to a “chemical oceanographer,” who is kind of like an oceanographer but specializing in the chemical composition of the oceans rather than their ecology, biological life and geology.
Example problems
1. Hydrofluoric acid reacts with calcium carbonate according to the following equation:
\(2 \mathrm{HF}(a q)+\mathrm{CaCO}_3 \rightarrow \mathrm{H}_2 \mathrm{O}(l)+\mathrm{CO}_2(g)+\mathrm{CaF}_2\)
If \(100 \mathrm{~mL}\) of a \(10 \mathrm{mM}\) solution of \(\mathrm{HF}\) is added to \(100 \mathrm{~mL}\) of a \(10 \mathrm{mM}\) solution of \(\mathrm{CaCO}_3\), how much \(\mathrm{CaF}_2\) will precipitate? Assume the reaction goes to completion.
- Answer
-
In this problem, the first clue is that the reaction "goes to completion:" this indicates that we don't need to worry about equilibrium. When a \(10 m M\) solution of \(\mathrm{HF}\) is added to a \(10 m M\) solution of \(\mathrm{CaCO}_3\), the final solution has a different concentration.
The original solutions each contain \(1 \mathrm{mmol}\) of material, since \(0.1 L * 10 \mathrm{mmol} / L=1 \mathrm{mmol}\). The final solution, therefore, has \(1 \mathrm{mmol} \mathrm{Hf}\) and \(1 \mathrm{mmol} \mathrm{CaCO}_3\) in \(0.2 \mathrm{~L}\) of water, so the concentration of the final solution is \(5 \mathrm{mM}\) in both reactants.
Next, we need to think about how much \(\mathrm{CaF}_2\) we can form: if we start with \(1 \mathrm{mmol}\) of \(\mathrm{HF}\) and 1 mmol \(\mathrm{CaCO}_3\), you can check that the limiting reagent is \(\mathrm{CaCO}_3\), since we need twice as much \(\mathrm{HF}\) as \(\mathrm{CaCO}_3\) to form \(\mathrm{CaF}_2\). We'll end up with \(0.5 \mathrm{mmol} \mathrm{CaF}_2\), and leave behind \(0.5 \mathrm{mmol} \mathrm{CaCO}_3\).
Next, we need to determine how much will dissolve. We can use an ICE table and the common ion effect. We start with \(0.5 \mathrm{mmol}\) of calcium ions in \(0.2 \mathrm{~L}=0.25 \mathrm{mM}\):
\[\begin{array}{c|c|c|c}
& \mathrm{CaF}_2 & \mathrm{Ca}^{2+} & 2 F^{-} \\
\hline \mathrm{I} & & 2.5 \mathrm{mM} & 0 \mathrm{mM} \\
\hline \mathrm{C} & & +\mathrm{x} & +2 \mathrm{x} \\
\hline \mathrm{E} & & 2.5+\mathrm{x} & 2 \mathrm{x}
\end{array} \nonumber\]\begin{aligned}
5.3 \times 10^{-9} &=(.0025+x)(2 x)^2 \\
x &=0.000649
\end{aligned}Therefore, \(0.649 \mathrm{mM}\) will be soluble, leaving \(2.5 \mathrm{mM}-0.649 \mathrm{mM}=1.85 \mathrm{mM} C a F_2\) will precipitate.
\[1.85 \mathrm{mmol} / L * 0.2 L=0.37 \mathrm{mmol} \nonumber\]
\[0.37 \mathrm{mmol} * 78.07 \mathrm{~g} / \mathrm{mol}=28.9 \mathrm{mgCaF} \mathrm{C}_2 \nonumber\]
Therefore, \(28.9 \mathrm{mg}\) of \(\mathrm{CaF}_2\) will precipitate.
Lecture 30: Bronsted-Lowry Acids/Bases and Neutralization
Summary
In this lecture, we started by considering what happens if you combine an acid and a base directly: for example, \(\mathrm{NaOH}(\mathrm{aq})+\mathrm{HCl}(\mathrm{aq}) \rightarrow \mathrm{H} 2 \mathrm{O}(\mathrm{l})+\mathrm{NaCl}(\mathrm{s})\). Each of these products, the water and the salt, find an equilibrium of their own. So the arrow goes both ways \(\mathrm{H}++\mathrm{OH}-\leftrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{l})\) and water gives the usual neutral ion concentrations of \(\mathrm{H}+\) and \(\mathrm{OH}-\). Salt also will have its equilibrium reaction to form ions \(\mathrm{Na}+\) and \(\mathrm{Cl}-\) in solution. Sometime the \(\mathrm{Na}+\) and \(\mathrm{Cl}-\) ions called "spectators" when in solution, since they don't participate in making the solution either acidic or basic. The reaction of an acid with a base to make water and salt is called a neutralization reaction.

Bronsted and Lowry defined acids and bases more broadly: a Bronsted-Lowry acid is anything that releases \(\mathrm{H}^{+}\)ions. A Bronsted-Lowry base is anything that accepts \(\mathrm{H}+\) ions. A "conjugate pair" is a nice way to keep track of the proton transfer that happens in acid/base chemistry. If we consider the generic reaction \(\mathrm{HA}+\mathrm{H}_2 \mathrm{O}\), where we label the A for "acid" and purposefully write the \(\mathrm{H}\) separately since we know as an acid it will be giving that \(\mathrm{H}\) up as a \(\mathrm{H}^{+}\)ion, the molecule left the other conjugate pair over after giving up the \(\mathrm{H}+\) ion into solution, \(\mathrm{A}-\), is called its conjugate pair. Similarly, if it reacted with \(\mathrm{H}_2 \mathrm{O}\), then the \(\mathrm{H}_2 \mathrm{O}\) gained the \(\mathrm{H}+\) to become \(\mathrm{H}_3 \mathrm{O}^{+}\), and those two molecules are conjugate pairs. According to Bronsted-Lowry, an acid base reaction is essentially just a proton transfer reaction.
A general way to write an acid mixing with a base would be: \(\mathrm{HA}+\mathrm{B} \rightarrow \mathrm{BH}^{+}+\mathrm{A}^{-}\) where here it is very clear that the acid transfers a proton to the base.

How do we know why and when an acid or base is "strong" vs. "weak?" This table lists common strong acids and bases. Acids other than these six are essentially all weak acids. The only common strong bases are the hydroxides of the alkali metals and the heavier alkaline earths (\(\mathrm{Ca}, \mathrm{Sr}\), and \(\mathrm{Ba}\); any other bases are likely to be weak. We can quantify why these molecules are considered "strong" by considering their acid dissociation constants. For \(\mathrm{HCl}\), the acid dissociation constant, or in other words the equilibrium constant for the acid, \(\mathrm{K}_a=\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{Cl}^{-}\right] /[\mathrm{HCl}]\) \(\approx 106\) - this is huge, and it means effectively full dissociation. A strong acid like \(\mathrm{HCl}\) is in fact strong because it fully dissociates. And the opposite is true too: if an acid fully dissociates, then it is a strong acid. The same applies for strong bases.
For example, suppose you have a \(1.0 \mathrm{M}\) solution of \(\mathrm{CH}_3 \mathrm{COOH}\). That means that there's 1 mole of \(\mathrm{CH}_3 \mathrm{COOH}\) in a liter of water. Since \(K_a=10^{-5}\), that means that \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\) for this \(1 \mathrm{M}\) solution is around \(0.003\), which in turn means that the \(\mathrm{pH}\) of \(1 \mathrm{M} \mathrm{CH}_3 \mathrm{COOH}\) is about \(2.5\). Note that if there's \(0.003\) moles of \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\) that can from 1 mole of \(\mathrm{CH}_3 \mathrm{COOH}\), then that means that only \(0.3 \%\) of it dissolved. That's not a lot of dissolution compared to the near \(100 \%\) for the strong acids!
Here's another example: will the salt formed from the following reaction have a \(\mathrm{pH}\) greater than, less than, or equal to \(7? \mathrm{CH}_3 \mathrm{COOH}(\mathrm{aq})+\mathrm{NaOH}(\mathrm{s}) \leftrightarrow \mathrm{Na}^{+}+\mathrm{CH}_3 \mathrm{COO}^{-}(\mathrm{aq})+\mathrm{H}_2 \mathrm{O}(\mathrm{l})\) To answer this we don't even need to do any math. That's because if a weak acid is mixed with a strong base (as is the case here), we automatically know that it will be basic.
In general, when the following are mixed, the results are:
- Weak acid mixed with weak base: \(\mathrm{pH}<7\) for \(K_a>K_b ; \mathrm{pH}=7\) if \(K_a=K_b ; \mathrm{pH}>7\) for \(K_a<K_b\)
- Strong acid mixed with string base: \(\mathrm{pH}=7\)
- Strong acid mixed with weak base: \(\mathrm{pH}<7\)
- Strong base mixed with weak acid: \(\mathrm{pH}>7\)
Why this matters
It might feel a bit on the hot side at \(470^{\circ} \mathrm{C}\), and you'd be surrounded by sulfuric acid most of the time. That's what it would be like if you lived on Venus. Here's a nice picture from a Russian space probe back in the 1970's that landed on the surface, showing what a stroll would be like over there. These temperatures and acidic atmospheres may sound crazy, but many materials require similar conditions for processing. A lot of aspects of our industrial revolution have effectively relied on reproducing the conditions of Venus! In fact, today, nearly 250 million tons of sulfuric acid is produced per year, which makes it one of the largest chemicals produced worldwide. The liberated \(\mathrm{H}+\) ions that come from \(\mathrm{H} 2 \mathrm{SO} 4\) are put to use in a wide range of industries, from detergents to lead-acid car batteries to dyes to metal processing, it is in the production of fertilizers where its use dominates: roughly \(50 \%\) of all sulfuric acid is used to make fertilizer!
In one of our earlier Why This Matters, we discussed the Haber-Bosch process, which uses a catalyst, high temperatures (like \(\approx 400^{\circ} \mathrm{C}\) ), and high pressures (like \(\approx 180\) atm.) to fix \(\mathrm{N}_2\) molecules and turn them into ammonia, \(\mathrm{NH}_3\). But the thing is, that's only one of the three most important ingredients plants need to grow: the other two are phosphorus and potassium. Phosphorus, in particular, is typically made with vast amounts of sulfuric acid. Why is this the case? You already know the answer, it's the chemistry.
found in sedimentary deposits, meaning that they formed by deposition of phosphate-rich materials in marine environments. Here's a picture of a phosphate rock formation being mined in Utah. But if you take phosphate rock and grind it up into a powder, it still won't be very soluble in water (we now have some knowledge about what that means!). This means that if you put the mineral directly into fertilizer, it will not be useful to plants. It's just like for the case of \(\mathrm{N}_2\) in the air: even though there's plenty of \(\mathrm{N}_2\), plants have no way of absorbing it. Rather, they need to work with \(\mathrm{NH}_3\) in order to take in nitrogen. The same thing is true for phosphorus: tricalcium phosphate, \(\mathrm{Ca}_3\left(\mathrm{PO}_4\right)_2\), which is what phosphate rock mostly consists of, is not able to give phosphorus to plants because it doesn't dissolve enough in water. The \(\mathrm{K}_{s p}\) for this ionic salt is a whoppingly low \(2.07 \times 10^{-33}\)! By the way, it's a pretty good thing that this doesn't dissolve very much since it's a key ingredient in bones and teeth.
That's where sulfuric acid comes in: it's a fairly simple way to liberate the phosphoric acid \(\mathrm{H}_3 \mathrm{PO}_4\) from the phosphate rock. In particular, the \(\mathrm{H}_2 \mathrm{SO}_4\) from the phosphate rock. In particular, the \(\mathrm{H}_2 \mathrm{SO}_4\) gives up its hydrogen in exchange for the calcium to form calcium sulfate, as follows:
\(\mathrm{Ca}_3\left(\mathrm{PO}_4\right)_2+3 \mathrm{H}_2 \mathrm{SO}_4 \rightarrow 2 \mathrm{H}_3 \mathrm{PO}_4+3 \mathrm{CaSO}_4\)
Once the phosphoric acid is produced in this manner, it can then be made into different types of fertilizer that are now water-soluble, so the plants can absorb it. These are generally termed "phosphates," or salts of phosphoric acid, which in the context of fertilizers include ammonium phosphates, calcium phosphates, and sodium phosphates. Bottom line: we feed the world because of our knowledge of acid/base chemistry!
Why this employs
Acids and bases are used so much in industry and in life: it's not hard to find jobs that involve acids and bases. You could check out DuPont, for example, which sells phosphate fertilizer plants that do just what we discussed above (for example, see http://cleantechnologies.dupont.com/ industries/phosphate-fertilizer/). http://cleantechnologies.dupont.com/...atefertilizer/. BASF was one of the very first companies to make and sell sulfuric acid on large scales, and they still do and have many job openings related to acid chemistry. There's Sigma Aldrich, which is a massive company with 10,000 employees and nearly \(\$ 3 \mathrm{~B}\) in annual revenue: their "acids and bases" category shows hundreds of products (btw, another company, Merck, bought them recently for \(\$ 17 \mathrm{~B})\). Or how about Cabot Laboratories, which doesn't make acids and bases explicitly but uses them to make every one of the cool chemical products they do make, whether elastomers or aerogels or "advanced carbons."
You could take any one of many different acids, look up its name, and likely find a market for it, and then all the companies that either make it or use it, many of which will have jobs related to it. Take formic acid: it’s getting more interest from a range of different industries because it’s easier to handle than other acids (see for example this article about it in C&EN (https://cen.acs.org/ articles/93/i48/Chemical-Makers-Eyes-Formic-Acid.html), and is used heavily in the textiles industry. BASF and Eastman Chemical are big on formic acid, but a lot of other companies are getting interested. Speaking of textiles, that’s another industry that uses massive amounts of acids, including formic acid but also citric, acetic, hydrochloric, and nitric acid. Textiles also rely heavily on bases like sodium hydroxide and baking soda for dyeing, cleaning, and fire-proofing clothes to name a few uses. All just to say that acids and bases and truly a bedrock of industry, and knowing how to make them or how to use them leads to massive job opportunities.
Example Problems
1. Identify the conjugate acid/base pairs in the following reactions:
a) \(\mathrm{H}_2 \mathrm{PO}_4^{-}+\mathrm{H}_2 \mathrm{O} \leftrightarrow \mathrm{HPO}_4^{2-}+\mathrm{H}_3 \mathrm{O}^{+}\)
- Answer
-

b) \(\mathrm{H}_2 \mathrm{O}+\mathrm{NH}_3 \leftrightarrow \mathrm{OH}^{-}+\mathrm{NH}_4^{+}\)
- Answer
-

2. Calculate the \(K_a\) of an \(0.2 \mathrm{M}\) aqueous solution of propionic acid \(\left(\mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{H}\right)\) with a pH of \(4.88\). The dissociation can be expressed as
\(\mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{H}+\mathrm{H}_2 \mathrm{O} \leftrightarrow \mathrm{H}_3 \mathrm{O}^{+}+\mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2^{-}\)
- Answer
-
This problem can be solved with an ICE table:
\[\begin{array}{c|c|c|c}
& \mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{H} & \mathrm{H}_3 \mathrm{O}^{+} & \mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2^{-} \\
\hline \mathrm{I} & 0.2 & 0 & 0 \\
\hline \mathrm{C} & -\mathrm{x} & +\mathrm{x} & +\mathrm{x} \\
\hline \mathrm{E} & 0.2-\mathrm{x} & \mathrm{x} & \mathrm{x}
\end{array} \nonumber\]\begin{gathered}
-p H=\log \left[H_3 O^{+}\right]=-4.88 \\
\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=10^{-4.88}=1.32 \times 10^{-5}=x \\
K_a=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2^{-}\right]}{\left[\mathrm{CH}_3 \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{H}\right]}=\frac{x^2}{0.2-x}=\frac{\left(1.32 \times 10^{-5}\right)^2}{0.2-1.32 \times 10^{-5}}=8.69 \times 10^{-10}
\end{gathered}
Further Reading
Lecture 21: Bragg’s Law and x-ray Diffraction
• More Bragg’s Law:
• How to do an XRD experiment:
https://serc.carleton.edu/research_education/geochemsheets/ techniques/XRD.html
• History of science: Rosalind Franklin, X-ray Diffraction, and the structure of DNA:
https: //doi-org.stanford.idm.oclc.org/10.1119/1.5020051
Lecture 22: From x-ray Diffraction to Crystal Structure
• Laue condition (beyond-the-scope of 3.091):
http://www.physics.udel.edu/~yji/PHYS624/ Chapter3.pdf
• Nice visualizations:
http://web.pdx.edu/~pmoeck/phy381/Topic5a-XRD.pdf
Lecture 23: Point Defects
• Schottky and Frenkel defects, plus some beyond-the-scope stuff:
http://ww2.chemistry.gatech. edu/class/6182/wilkinson/nonstoi.pdf
• How defects give different gemstones their distinctive looks:
https://www.tf.uni-kiel.de/ matwis/amat/iss/kap_6/advanced/t6_1_1.html
Lecture 24: Line Defects and Stress-Strain Curves
• Defects summary:
https://chemed.chem.purdue.edu/genchem/topicreview/bp/materials/ defects3.html
• Animation of moving edge dislocation:
https://www.tf.uni-kiel.de/matwis/amat/def_en/ kap_5/illustr/a5_1_1.html
• Video on stress-strain curves:
Lecture 25: Amorphous Materials: Glassy Solids
• Crystalline vs. amorphous solids:
• Record for image of thinnest glass:
https://news.cornell.edu/stories/2013/09/shattering-records-thinnest-glass-guinness-book
Lecture 26: Engineering Glass Properties
• Inside a Corning factory:
https://www.youtube.com/watch?v=gZPeyErbqz4
• 3D printing glass:
https://www.youtube.com/watch?v=IvcpbtpWpGY
Lecture 27: Reaction Rates
• Summary of reaction rates:
• More on catalytic converters:
Lecture 28: Equilibrium and solubility
• Solubility product practice:
https://www.chem.purdue.edu/gchelp/howtosolveit/Equilibrium/ Solubility_Products.htm
• A method for making more medicines soluble:
Lecture 29: Common Ions and Acids/Bases
• Worked out common ion effect example:
• Video on pH:
Lecture 30: Bronsted-Lowry Acids/Bases and Neutralization
• The Bronsted concept of acids and bases:
• Acids and bases in cooking:
https://www.livestrong.com/article/431096-acids-bases-in-cooking/