
2.5: Pi Coordination- Donation from Alkenes
- Page ID
- 189929

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Lone pairs are the most common electron donors in coordination complexes. Chloride ions, ammonia and phosphines all donate a lone pair to metals to form complexes. Lone pairs are not stabilized by bonding interactions already. Forming a bond lowers the energy of the lone pair electrons. On the other hand, bonding pairs are already lower in energy and so they are less likely to be donated.
However, pi bonds can also donate to Lewis acids. Donation from sigma bonds is still much less likely, though. Sigma bonds are buried between atoms and are hard to reach.
- Pi bonds are often higher in energy than corresponding sigma bonds
- Pi bonds are more accessible spatially than sigma bonds
A simple example of donation from a pi bond is the treatment of silver(I) salts with alkenes, shown in figure \(\PageIndex{1}\).

Strictly speaking, if an alkene donates its pi bonding electrons to a metal, we might draw it as shown in Figure CC4.2. The electrons are shared between two carbon atoms in the alkene. Since a bond is generally thought of as a pair of electrons shared between two atoms, then once the pi electrons are donated to the metal, they are shared between the metal and one of the carbons, but not both. If you are keeping track of electrons carefully, that leaves one of the carbons short on electrons. It must be a cation. Of course, either carbon could be the cationic one, so we can draw resonance structures showing both possible states.
- donation of pi electrons to a metal can "activate" an alkene
- the alkene can become positive and electrophilic
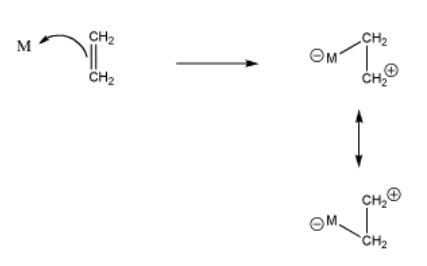
However, if a metal has valence electrons of its own, it could donate these electrons back to the "cation" that is forming on one of the alkene carbons. The alkene complex can be thought of as a "metallacycle" or a "metallacyclopropane", a three-membered ring containing two carbons and the transition metal atom.
- alkene coordination also involves metal-to-alkene donation
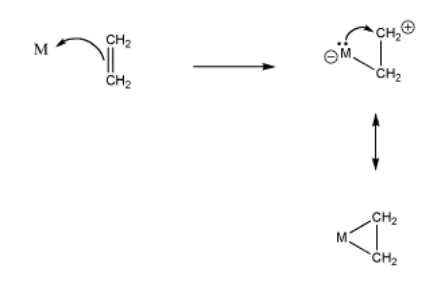
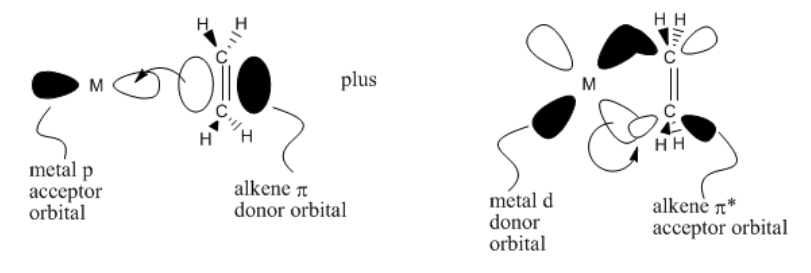
The idea of back-donation is also supported from a molecular orbital point of view. The alkene pi bond can donate electrons into an empty orbital on the metal, such as a p orbital. In turn, an occupied metal d orbital has the correct symmetry to overlap with a pi* orbital on the alkene. In doing so, we would think of the pi bond as breaking. We would also think of two pairs of bonding electrons between the metal and alkenes. This situation fits the picture of a metallacyle pretty well.
- alkene-to-metal donation and metal-to-alkene donation are supported by molecular orbital calculations
- without both components, alkenes do not bind very well to metals
- nevertheless, the sigma bond formed by donation from the alkene is still the principle bond-forming event.
Remember that formalisms can be complicated in coordination complexes. For one thing, we do not usually draw positive formal charges on the donor atom or negative formal charges on the metal atom in the complex (unless specifically illustrating a point). In alkene complexes, bonding is usually illustrated with a line between the pi bond and the metal, as in Figure CC4.5. That line could be read as a pair of electrons, but it isn't, really. The pair of electrons is in the pi bond. They are being shared with the metal.
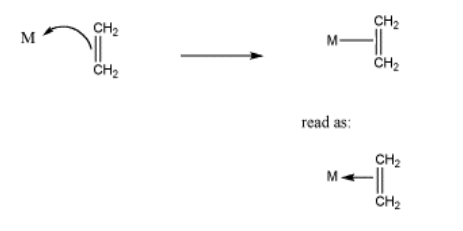
Figure \(\PageIndex{5}\): Typical representation of alkene complexes.
Alkene binding is one of the first steps performed by hydrogenation catalysts such as Wilkinson's catalyst, which catalyze the addition of dihydrogen across an alkene double bond to form an alkane.
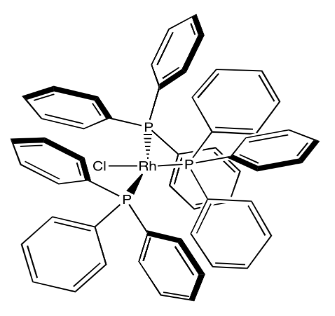
Show, with arrows, the coordination of cyclohexene to Wilkinson's catalyst, (PPh3)3RhCl.
- Answer
-

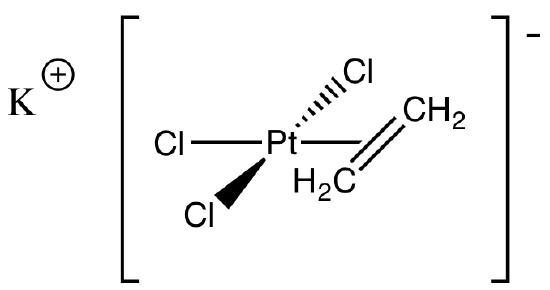
The first example of an alkene coordinated to a transition metal was prepared by pharmaceutical chemist W. C. Ziese at the University of Copenhagen in 1827. Its structure was confirmed by x-ray diffraction about a century later. Its formula is K[PtCl3(CH2CH2)]. Draw the structure.
- Answer
-
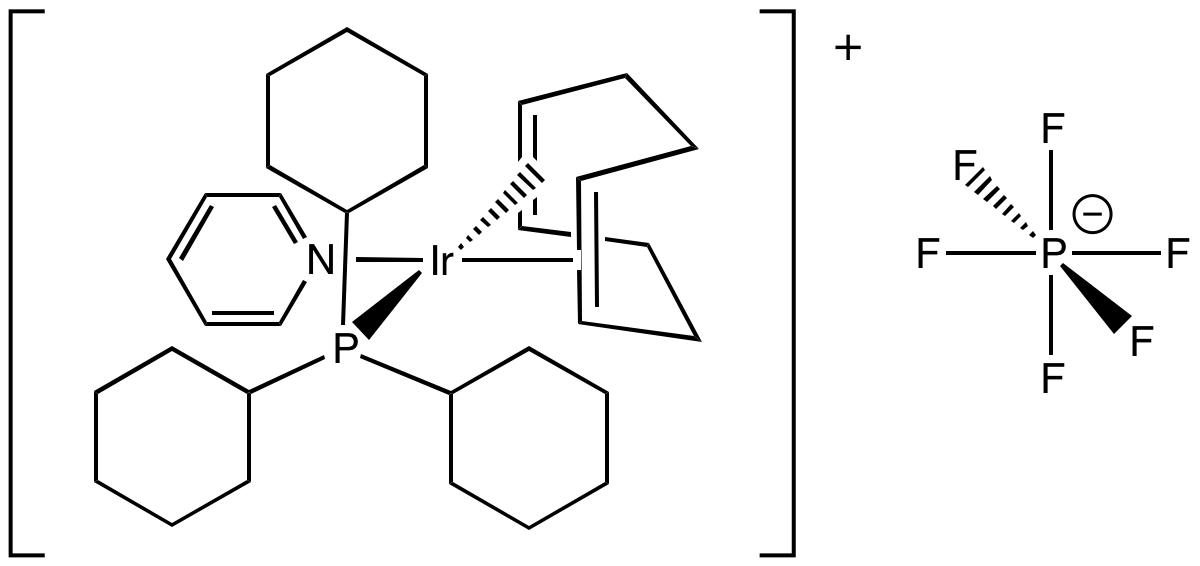
Crabtree's catalyst is a hydrogenation catalyst with formula [(COD)(py)(PCy3)Ir] PF6. Note that COD = 1,4-cyclooctadiene; py = pyridine; Cy = cyclohexyl. Draw the structure of this square planar iridium complex.
- Answer
-
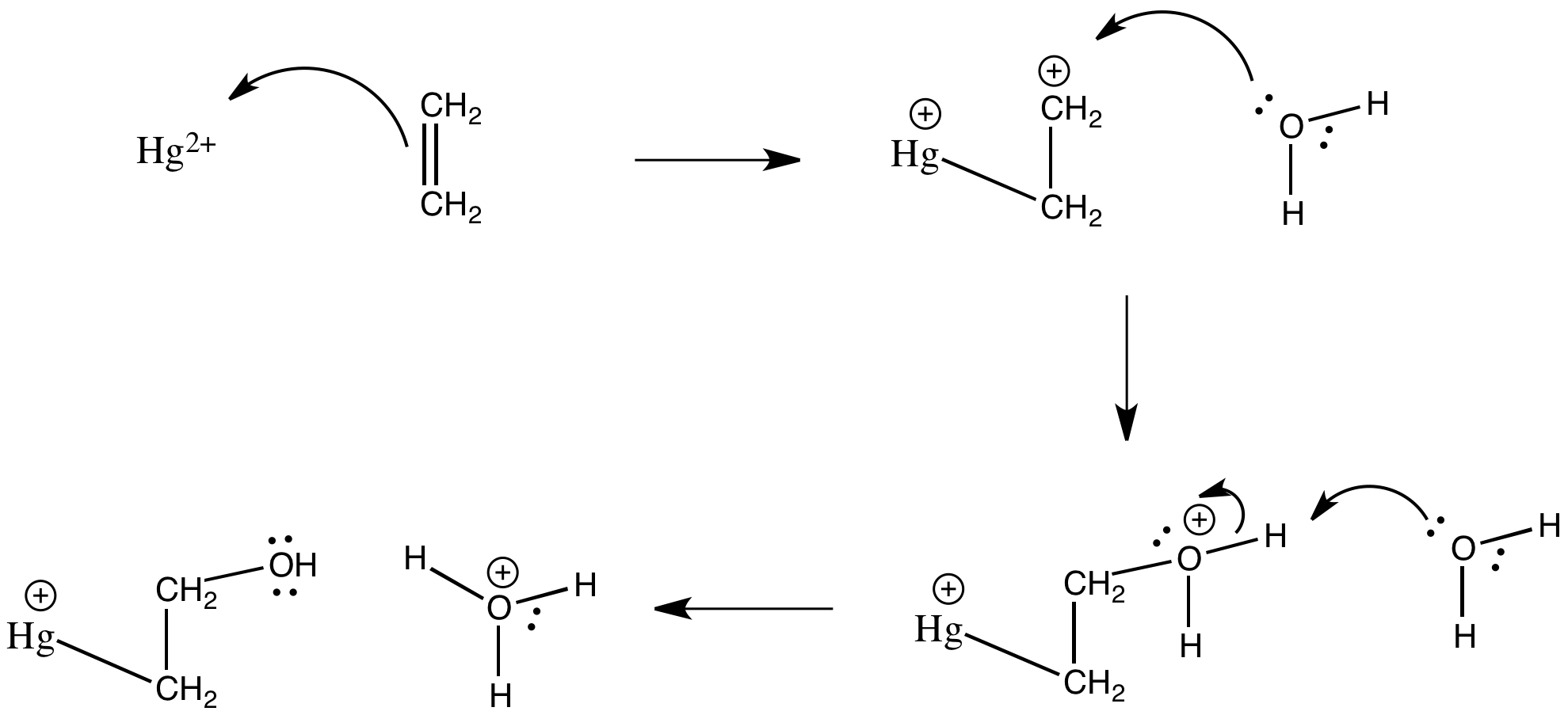
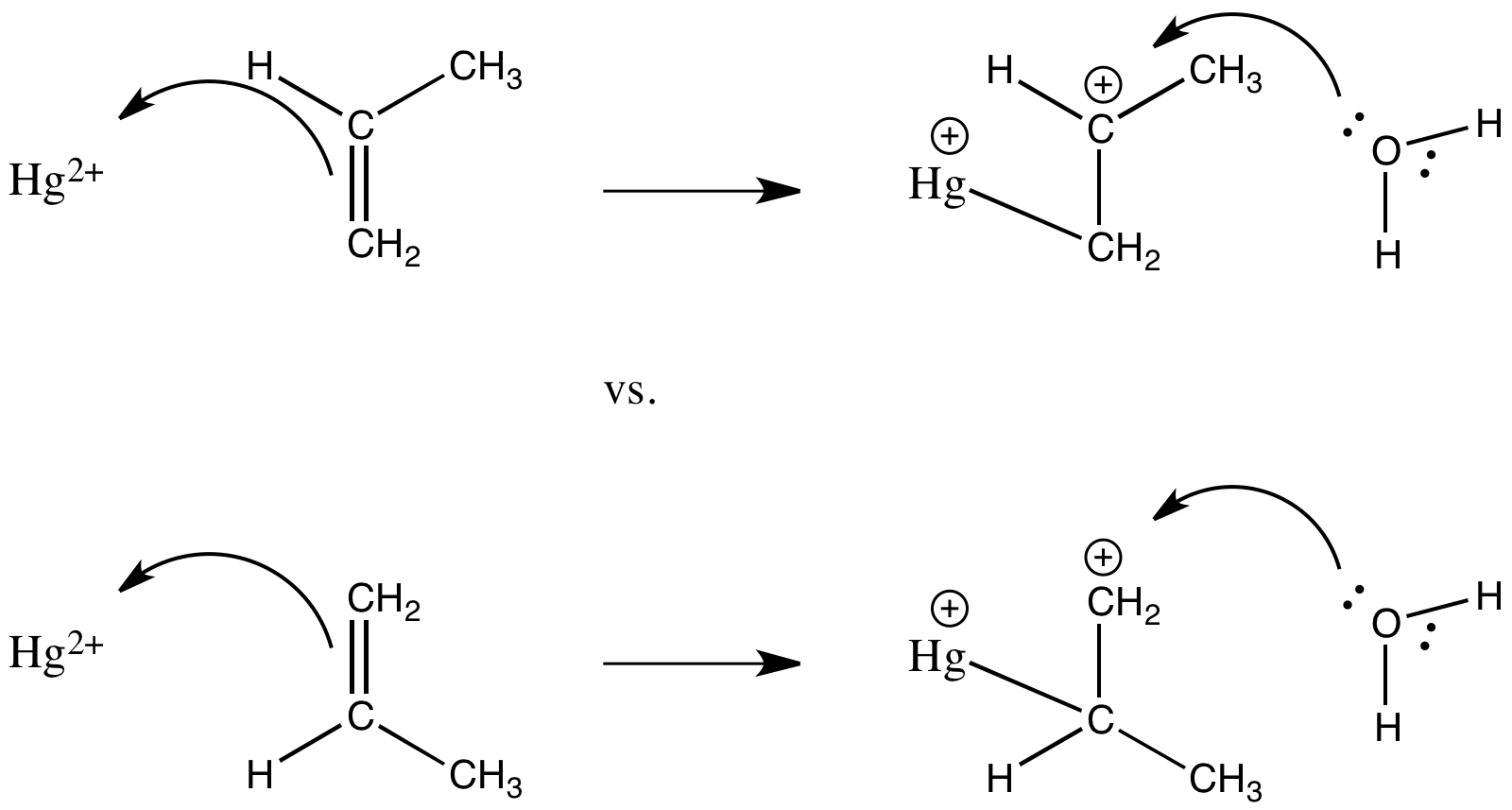
Treatment of alkenes with Hg(II) in water results in addition of a solvent molecule (a nucleophile) to one end of the "activated" alkene. Draw, with arrows, the mechanism for the formation of a hydroxyethylmercury ion, HgCH2CH2OH+, from ethene under these conditions.
- Answer
-
Treatment of 2-methylpropene with Hg(II) in water results in formation of the ion, HgCH2C(CH3)2OH+. A second product of solvent addition is possible, but is not observed. Show the other possible product and provide a possible explanation for the selectivity of the reaction.
- Answer
-
Alkenes coordinate to many metals tightly enough that alkene complexes can be isolated and characterized. However, although early metal ions such as Zr(IV) are believed to bind alkenes, they do not coordinate tightly enough to form stable compounds that can be isolated and characterized. Explain why.
- Answer
-
Zr(IV) or Zr4+ has no valence d electrons. That means that, although an alkene could certainly donate its pi bond to the zirconium atom, the zirconium has no electrons with which it can stabilize the alkene complex via "back-donation" to the pi antibonding orbital on the alkene.
Nevertheless, d0 metals such as Zr(IV) and Ti(IV) can be used as alkene polymerization catalysts to make common plastics such as HDPE, LDPE and polypropylene. That means that, although an alkene complex isn't directly observed with these metal ions, these metals can evidently bind alkenes briefly and get them to react with other alkenes to form long chains. Even so, most industrial olefin polymerization catalysts use Ti(III).
Alkynes can also coordinate to metal atoms. Draw the molecular orbitals involved in:
a) alkyne donation to the metal
b) metal donation to the alkyne
- Answer
-
Add texts here. Do not delete this text first.
Explain the differences seen in the equilibrium constants for the formation of silver(I) complexes with the following alkenes:
a) CH2CH2: K = 22.3
b) cis-CH3CHCHCH2CH2CH3: K = 3.1
c) trans-CH3CHCHCH2CH2CH3: K = 0.8
- Answer
-
We will use (a), the binding constant between Ag(I) and ethylene or ethene (CH2=CH2), as our baseline value. Other constants will be compared with this one in order to look for a trend.
In (b), the binding constant is much smaller, so the silver ion binds cis-2-hexene much less tightly than it does ethene. This is just another alkene, like ethene, but instead of having just hydrogen atoms attached to the C=C unit, cis-2-hexene has some other stuff. Maybe this other stuff causes some problem for alkene binding. The obvious difference between hydrogen atoms and this other stuff is that this other stuff is bigger. Maybe the complex gets too crowded when the cis-2-hexene binds to the silver ion.
Very often when we see metal ions on paper, we are not dealing with bare metal ions in reality. The ion often has other ligands already attached to it at the beginning, such as water molecules, and what we are sometimes looking at is replacement of an old ligand with a new ligand. Other ligands attached to the silver could make crowding problems even worse.
There is an alternative explanation as well. If you compare two alkenes that differ only in the number of hydrogens attached to the double bond, such as 1-butene, CH2=CHCH2CH3, and 2-butene, CH3CH=CHCH3, you invariably find that the alkene with fewer hydrogens attached to the double bond (and more other stuff) is more stable. "Terminal alkenes", with double bonds at the ends of the chain, are always less stable than "internal alkenes", with double bonds somewhere along the middle of the chain. This difference can be explained by looking at some quantum mechanical calculations, but we're not going to do that right now.
The point is, the difference in this reaction might be caused, not by the alkene complexes, but by the alkenes themselves, on the other side of the reaction profile. It's important to remember that equilibrium constants always compare two sides of a reaction. Ethene, having fewer substituents on the double bond than cis-2-hexene ("substituents" is a four syllable word for "other stuff"), may simply be less stable and more reactive.
Do either of these ideas hold up in the other examples?
In (c), trans-2-hexene is bound even less tightly than cis-2-hexene. We could argue that in a cis-2-hexene complex, the substituents, which are on the same side of the double bond, might both be held away from other ligands on the metal that may exacerbate crowding problems. That would be more difficult to do with trans-2-hexene, since one substituent is on either side of the double bond. Getting one substituent away from the crowding may be possible, but probably not both.
Once again, the alternative explanation holds up here, too. cis-2-Hexene is less stable than trans-2-hexene, because the substituents on the double bond crowd each other in cis-2-hexene, but are held away from each other in trans-2-hexene. So maybe cis-2-hexene binds to silver ion more easily because it is more reactive.