Anticancer Drugs
- Page ID
- 89057
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)When fighting cancer, the entire population of neoplastic cells must be eradicated in order to obtain desired results. The concept of "total cell-kill" applies to chemotherapy as it does to other means of treatment: total excision of the tumor is necessary for surgical care, and complete destruction of all cancer cells is required for a cure with radiation therapy. By investigation of a model tumor system, the L1210 leukemia of mice, a number of important principles have been established as follows:
- A single clonogenic malignant cell can give rise to sufficient progeny to kill the host; to achieve cure it is thus necessary to destroy every such cell. Since the doubling-time of most tumors is relatively constant during logarithmic growth, the life-span of the host is inversely related to the number of malignant cells that are inoculated or that survive therapeutic measures.
- In contrast to antimicrobial chemotherapy where, in most instances, there are major contributions by the immune mechanisms and other host defenses, these play a negligible role in the therapy of neoplastic disease unless only a small number of malignant cells is present.
- The cell-kill caused by antineoplastic agents follows first-order kinetics, that is, a constant percentage, rether than a constant number, of cells is killed by a given therapeutic maneuver, this finding has had a profound impact on clinical cancer chemotherapy. For example, a patient with advanced acute lymphocytic leukemia might harbor 1012 or about 1 kg of malignant cells. A drug killing 99.99% of these cells would reduce the tumor mass to about 100mg, and this would be apparent as a complete clinical remission. However, 108malignant cells would remain, any of which could cause a relapse in the disease.
The logical outgrowth of these concepts has been the attempt to achieve total cell-kill by the use of several chemotherapeutic agents concurrently or in rational sequences. The resulting prolonged survival of patients with acute lymphocytic leukemia through the use of such multiple-drug regimens has encouraged the application of these principles the treatment of other neoplasms.
Fundamental advances continue in the chemotherapy of neoplastic diseases. The greatest progress in recent years has not been the discovery of new, useful chemotherapeutic agents but at the conceptual level: the design of more effective regimens for concurrent administration of drugs; the acquisition of knowledge of the mechanisms of action of many antitumor agents, which facilitates the design of new methods to prevent or minimize drug toxicity; the increased use of adjuvant chemotherapy (e,g., the design of chemotherapeutic approaches to destroy micrometastases and prevent the development of secondary neoplasms after removal of destruction of the primary tumor by surgery of irradiation); and increased knowledge about such vital processes as tumor initiation and the dissemination, implantation, and growth of metastases. Of great importance is recognition of the problems imposed by the heterogeneity of tumors, with the realization that individual tumors may contain many subpopulations of neoplastic cells that differ in crucial characteristics, such as karyotype, morphology, immunogenicity, rate of growth, the capacity to metastasize, and , significantly, responsiveness to antineoplastic agents. Information also continues to accumulate in the fields of molecular and cellular biology, resulting in a greater understanding of cellular division and differentiation, tumor immunology, and viral and chemical carcinogenisis. It is hoped that these discoveries will provide new targets for therapy.
A. Alkylating Agents
The chemotherapeutic alylating agents have in common the property of undergoing strongly electrophilic chemical reactions through the formation of carbonium ion intermediates or of transition complexes with the target molecules. These reactions result in the formation of covalent linkages (alkylation) with various nucleophilic substances, including such biologically important moieties as phosphate, amino, sulfydryl, hydroxyl, carbonyl, and imidozole groups. The cytotoxic and other effects of the alkylating agents are directly related to the alkylation of components of DNA. The 7 nitrogen atom of guanine is particularly susceptible to the formation of a covalent bond with both monofunctional and bifunctional alkylators and may well represent the key target that determines the biological effects of these agents. It must be appreciated, however, that other atoms in the purine and pyrimidine bases of DNA-for example, the 1 or 3 nitrogens of adenine, the 3 nitrogen of cytosine, and the 6 oxygen of guanine-may also be alkylated to a lesser degree, as are the phosphate atom of the DNA chains and the proteins associated with DNA.
Nitrogen Mustards
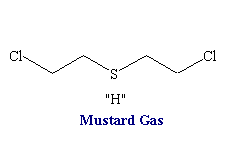
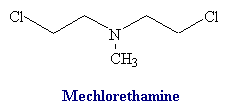
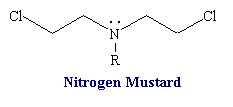
Structure of nitrogen mustards. 3D structure of nitrogen mustard and uracil nitrogen mustard
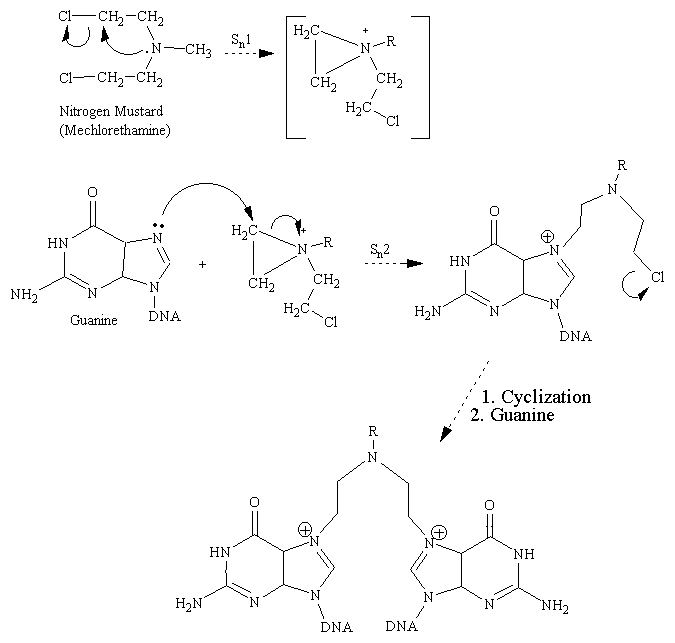
Alkylating Mechanism of Mechlorethamine With Guanine Base:
Cyclosphosphamide
Efforts to modify the chemical structure of mechlorethamine to achieve greater selectivity for neoplastic tissues led to the development of cyclophosphamide. After studies of the pharmacological activity of cyclophosphamide, clinical investigations by European workers demonstrated its effectiveness in selected malignant neoplasms.
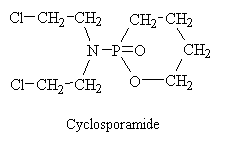
Cyclophosphamide is a classical example of the role of the host metabolism in the activation of an alkylating agent and is one or the most widely used agents of this class. The original rationale that guided its molecular design was twofold. First, if a cyclic phosphamide group replaced the N-methyl of mechlorethamine, the compound might be relatively inert, presumably because the bis-(2-chloroethyl) group of the molecule could not ionize until the cyclic phosphamide was cleaved at the phosphorous-nitrogen linkage. Second, it was hoped that neplastic tissues might posses phosphatase of phosphamidase activity capable of accomplishing this cleavage, thus resulting in the selective production of an activated nitrogen mustard in the malignant cells. In accord with these predictions, cyclophosphamide displays only weak cytotoxic, mutagenic, or alkylating activity and is relatively stable in aqueous solution. However, when administered to experimental animals or patients bearing susceptible tumors, marked chemotherapeutic effects, as well as mutagenicity and cancinogenicity, are seen. Although a definite role for phosphatases of phosphamidases in the mechanism of action of cyclophospamide has not yet been demonstrated, it is clearly established that the drug initially undergoes metabolic activation be the cytochrome P-450 mixed-function oxidase system of the liver, with subsequent transport of the activated intermediate to sites of action. Thus, a crucial factor in the structure-activity relationship of cyclophosphamide concerns its capacity to undergo metabolic activation in the liver, rather than to alkylated malignant cells directly. it also appears that the selectivity of cyclophosphamide against certain malignant tissues may result in part from the capacity of normal tissues, such as liver, to protect themselves against cytotoxicity by further degrading the activated intermediates.
None of the severe acute CNS manifestations reported with the typical nitrogen mustards has been noted with cyclophosphamide. Nosea and vomiting, however, may occur. Although the general cytotoxic action of this drug is similar to that of other alkylating agents, some notable diferences have been observed. When comapred with mechloroethamine, damage to the megakaryocytes and thrombocytopenia are less common. Another unusual manifestation of selectivity consists in more prominent damage to the hair follicles, resulting frequently in alopecia (baldness). The drug is not a vesicant, and local irritaion does occur.
Uracil Mustard
Uracil mustard was synthesized in an unsuccessful attempt to produce an active-site alkylator by linking the bis-(2-chloroethyl) group to the pyrimidine base uracil. Its activity in experimental neoplasms was demonstrated shortly thereafter. No relationship has been demonstrated, however, with the biological function of uracil. Note: side effects of chemotherapy can be treated with marijuana
B. Anti-metabolites - (pyrimidine antagonists)
Cancer is a group of diseases characterized by abnormal and uncontrolled cell division. One important approach to antitumor agents is the design of compounds with structures related to those of pyrimidines and purines that are involved in biosynthesis of DNA. These compounds are known as antimetabolites because they interfere with the formation or utilization of a normal cellular metabolite. This interference generally results from the inhibition of an enzyme in the biosynthetic pathway of the metabolite from the incorporation, as a false building block, into vital macromolecules such as proteins or nucleic acids.
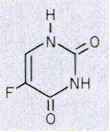
5-Fluorouracile - Pyrimidine Antagonist
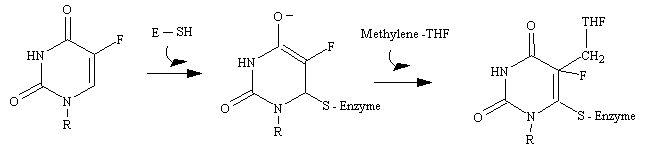
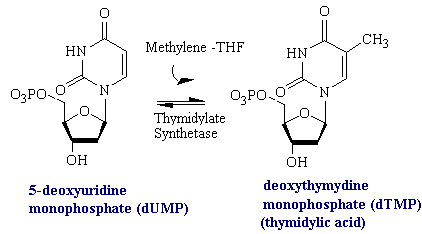
Uracil is not a component of DNA. Rather, DNA contains thymine, the methylated analog of uracil. The enzyme thymidylate synthetase is required to catalyze this finishing touch: deoxyuridylate (dUMP) is methylated to deoxythymidylate (dTMP) (see figure below). The methyl donor in this reaction is methylenetetrahydrofolate. Rapidly dividing cells require an abundant supply of deoxythymadylate for the synthesis of DNA. Therefore, the vulnerability of these cells to the inhibition of dTMP synthesis can be exploited in cancer therapy.
The rational for 5-fluorouracil, 5-FU, was to block DNA synthesis by inhibiting the biosynthesis of dTMP, by virtue of its close structural analogy to uracil. Fluorine, being the smallest atom that would substitute for hydrogen at the 5' position, was assumed to create the smallest possible molecular perturbation and thus be converted to the nucleotide and be accepted by the reactive site of thymidylate synthetase as a substrate imposter. In fact, this was the case. The van der Waals radius of the F atom (1.35 A) is only slightly larger than that of the H atom (1.20 A). Therefore, 5-fluorouracil is a fluorinated pyrimidine analogue which stops cell proliferation by blocking DNA synthesis and RNA processing.
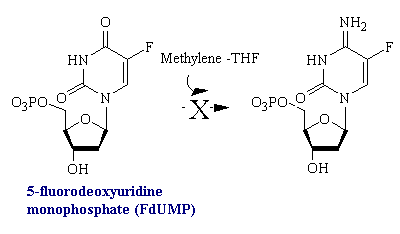
Fluorouracil is converted in vivo into fluorodeoxyuridylate (F-dUMP). This analog of dUMP irreversibly inhibits thymidylate synthase after acting as a normal substrate through part of the catalytic cycle. First a sulfhydryl group of the enzyme adds to C-6 of the bound F-dUMP (see figure below). Methylenetetrahydrofolate then adds to C-5 of this intermediate. In the case of dUMP, a hydride ion of the folate is subsequently shifted to the methylene group, and a proton is taken away from C-5 of the bound nucleotide. However, F+ cannot be abstracted from F-dUMP by the enzyme, and so catalysis is blocked at the stage of the covalent complex formed by F-dUMP, methylenetetrahydrofolate, and the sulfhydryl group of the enzyme. We see here and example of suicide inhibition, in which an enzyme converts a substrate into a reactive inhibitor that immediately inactivates its catalytic activity.