
Ion-Exchange Chromatography
- Page ID
- 71833

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A second sub-category of liquid chromatography is known as ion-exchange chromatography. This technique is used to analyze ionic substances. It is often used for inorganic anions (e.g., chloride, nitrate, and sulfate) and inorganic cations (e.g., lithium, sodium, and potassium). It can also be used for organic ions, although this is less common with the advent of reversed phase liquid chromatographic methods that will be described later. Another significant application of ion-exchange chromatography is as a step in the purification of proteins. Some of the substituent groups of amino acids are charged (the total charge for a particular protein is a function of the pH of the solution), which makes ion exchange a suitable method for protein purification.
The approach is to attach fixed ionic groups to the surface of a solid support. One common support used in the formation of ion exchange resins is polystyrene-divinylbenzene copolymers. The fixed ions are attached through a derivatization of the phenyl rings of the polystryene. Common fixed ions involve either sulfonate groups or quaternary amines as shown in Figure 56. The aromatic sulfonate groups are strong enough acids that they are deprotonated at all but highly acidic pH values (pH < 1).
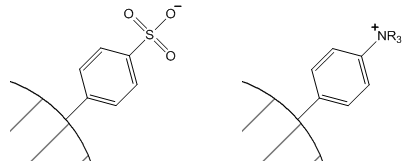
Sulfonate group Quaternary amine
Figure 56. Polystyrene polymer derivatized with fixed ionic groups.
Since we must preserve neutrality in such a system, there must also be an exchangeable counterion associated with each of these fixed groups. In the case of the sulfonate group, the counterion is a cation and this is a cation exchange resin. With the quarternary amine phases, the exchangeable counterion is an anion and this is an anion exchange resin. These counterions can be exchanged with each other. For example, this would enable you to have a cation exchange resin in the sodium form or the hydrogen form as seen in Figure 57
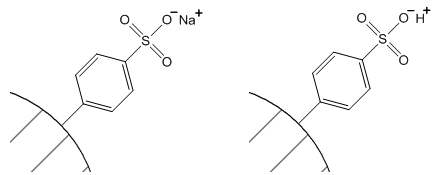
Sodium form Hydrogen form
Figure 57. Sulfonate cation exchange resin with sodium and hydrogen counterions.
An anion exchange resin could be in the hydroxide form (OH–) or chloride form (Cl–). One common use of ion exchange resins is in the deionization of water. It is useful to consider a scheme using ion exchange resins that would enable you to deionize water. If for example, we had water with sodium chloride in it (Na+Cl–), we would need a way of removing both cations and anions. If we first passed the water through a cation exchange resin in the hydrogen form, the sodium ions would exchange with the hydrogen ions as shown in the top picture of Figure 58. If we then passed the water through an anion exchange resin in the hydroxide form, the chloride ions would exchange with the hydroxide ions as shown in the bottom picture of Figure 58.
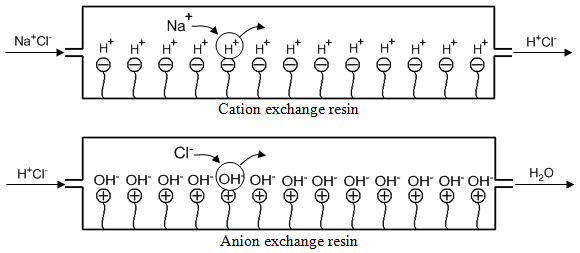
The H+ and OH– given off by the two resins would combine to form water. Eventually the resin will fill up with impurity ions (Na+ and Cl– in this case), and we would either need to replace the resin or reactivate it. We can reactivate the cation exchange resin by passing relatively concentrated hydrochloric acid through it to remove all the Na+ and replace it with H+. We can reactivate the anion exchange resin by passing a relatively concentrated solution of sodium hydroxide through it to remove all the Cl– and replace it with OH–. To minimize how often we need to replace these resins or how frequently we need to recharge them, it’s best to have resins with as high an ion exchange capacity as possible (the capacity is determined by the number of phenyl rings that have been derivatized with fixed ions).
The capacity of ion exchange resins used for deionizing water is too high for analytical purposes. Presumably, the concentrations of ions in samples that we want to analyze are relatively low. If we use high capacity resins, the retention times will be much too long. One of the early impediments to the use of ion exchange as an analytical method was the lack of methods to reproducibly synthesize resins with low capacities. One of the first groups of people to figure out a way to do this was chemists at Dow Chemical. They were motivated by a need to measure inorganic anions and cations at low levels and realized that ion-exchange chromatography would be an ideal method for doing so.
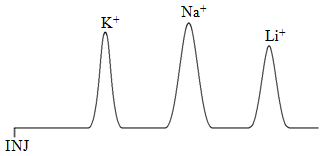
With any chromatographic method, it helps to know some basic rules for predicting retention order. An interesting example to consider in ion-exchange chromatography is the retention order for the ions Li+, Na+, and K+ on a cation exchange resin. One thing we can consider is whether these ions have different strength interactions with the fixed anion in the resin. All three ions have the same charge. What should also be apparent is that lithium is the smallest ion with the densest charge, potassium the largest with the most diffuse or fluffiest charge. We therefore might predict that the attraction of the lithium ion for the sulfonate groups in the resin is the strongest of the three ions. It turns out that the equation for ionic attraction has a distance term in it, and the closer the ions, the stronger the attraction. Lithium being the smallest is effectively closer to the sulfonate and has the strongest attractive force of the three for the resin. This would suggest a retention order with potassium eluting first, sodium second, and lithium last as shown in Figure 59.
But there is also a mobile phase in chromatographic separations, and it is useful to examine whether these ions have different relative stabilities in the mobile phase. If one of the three is most stable in the mobile phase, it might be expected to stay in the mobile phase more than the others and elute first. What might affect the stability of these ions in the mobile phase? The mobile phase in this separation would be an aqueous phase. If we consider water, we know that it is unusual in having a very elaborate network of hydrogen bonds. Any ion that dissolves in water will cause some disruption of this network. The question to consider is which ion would be the most disruptive of the three to the hydrogen bond network? Since the charges of these three ions are identical, this decision will be based on the size. The larger potassium ion will be more disruptive of the hydrogen bonds, and more difficult for water to accommodate. You could then say that Li+ is more stable in the water, stays in the mobile phase more, and elutes first. You could phrase this another way by saying that the K+ is least stable in the aqueous mobile phase, and so is “forced” into the resin by the mobile phase and elutes last. Sometimes the chromatographic literature refers to this as the solvophobic effect. The K+ fears the solvent and so spends more time in the resin. The relative retention order based on mobile phase effects would be lithium first and potassium last, as shown in Figure 60, the exact opposite of what was predicted above based on stationary phase effects.
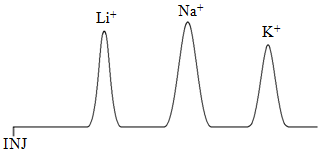
With conflicting predictions, the only way to know which one is more important is to inject the ions and determine the retention order. In this case, the measurements show that the Li+ elutes first and the K+ elutes last. The mobile phase effects are more significant in determining the retention order.
Now suppose we had two ions with exactly the same size, but one had a charge of 1+ and the other a charge of 2+. Examining stationary phase effects, the equation that describes the attraction between two ions has the charges of both ions in it. The greater the charge of the ions, the greater the attraction. We would therefore expect the 2+ ion to show greater attraction for the fixed sulfonate anions and elute later than the 1+ ion as shown in Figure 61.
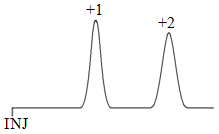
Examining mobile phase effects, both ions would have a similar extent of disruption of the hydrogen bond network since both have the same size. What we then need to consider is the strength of the attraction between the positive ion and the sphere of water molecules that surround it (remember, these cations would be surrounded by the slightly negative oxygen atoms of water molecules). This represents an electrostatic attraction, and again is dependent on charge. Therefore the 2+ ion is more stable in the water and ought to elute first as shown in Figure 62, the exact opposite of what was predicted based on stationary phase effects.
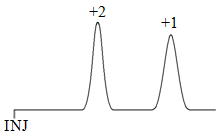
Faced with conflicting predictions, it’s necessary to perform the experiment and see which comes out first. In this case, the measurements show that the 1+ ion elutes first, the 2+ ion elutes second. The stationary phase effects are more important in this case in determining the retention order. The likely reason why the stationary phase effects are more significant is that the 2+ ion can actually bind simultaneously at two adjacent sulfonate sites as shown in Figure 63.
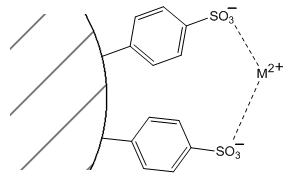
The likelihood that this occurs can be observed if a lanthanide(III) ion is added to these resins. In this case, the resin particles actually shrink in size as the lanthanide ion is added. The reason for the compression is that the binding of three sulfonate groups to the lanthanide causes the polymer to collapse in a bit to fit these groups around the lanthanide.
The separation of Li+, Na+, and K+ described in the prior problem would often be done on a polystyrene resin using a fairly dilute solution of hydrochloric acid (perhaps 0.1 M) as the mobile phase. The bound ions would be sulfonate groups and the mobile counter ion would be the H+ ion. An important issue is how to detect these ions. They do not absorb ultraviolet or visible light in the accessible portion of the spectrum. They do not absorb infrared light. Conductivity is one possibility for performing the measurement. The conductivity of a solution is a measure of the extent to which the solution conducts electricity. Dissolved ions are needed for a solution to conduct electricity, and the higher the concentration of ions, the higher the conductivity. We can measure the conductivity of a solution quite sensitively. In fact, this is the reading that is performed on water that has been purified by passage through a MilliQ water purification device to see just how well the water has been deionized. The only problem with trying to apply a direct conductimetric measurement is that the hydrochloric acid in the mobile phase produces too high a background signal. The chemists at Dow who had developed low capacity ion exchange resins recognized this problem as well and devised an ingeneous way to remove the conductivity of the eluent ions (HCl) but retain the conductivity of the alkali ions they wanted to detect.
What they did was use a device called a suppressor column. If we imagine measuring Na+ in a solution containing sodium chloride (Na+Cl–), we first start with a cation exchange resin in the H+ form and would have Na+Cl– and H+Cl– eluting out the end of the column when the sodium band comes off as shown in Figure 64.
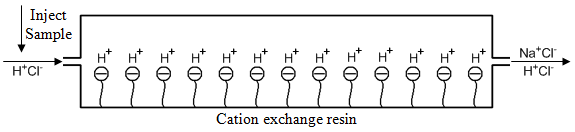
Note that there is still a high concentration of HCl mixed with the NaCl as it elutes from the column. This HCl will interfere with the measurement of conductivity. Suppose we took the column eluent and then passed it through an anion exchange resin in the hydroxide form as shown in Figure 65. The Cl– ion of the Na+Cl– would exchange with the hydroxide, converting this into sodium hydroxide (Na+OH–), a conducting electrolyte because it stays ionized. The Cl– ion of the HCl would exchange with the hydroxide, converting this into H+ and OH–, which is non-conducting water. We can therefore measure a conductivity that only relates to the amount of sodium ion in the original sample. An analogous scheme, which had the columns reversed, could be used to measure the conductivity of anions that were separated on an anion exchange column in the hydroxide form.
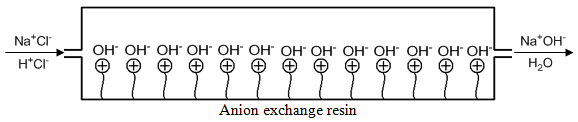
Eventually, the hydroxide counterions in the suppressor column will all become replaced with chloride ions and the device would not work anymore. The suppressor column must be periodically regenerated in the hydroxide form.
Today, instead of using suppressor columns to remove the conductivity of the eluent ions, membrane-based electrolytic neutralization devices are employed. The electrolysis of water can be used to generate hydronium and hydroxide ions, and by proper design, the desired ion can be generated in such a way to pass through a membrane and suppress the conductivity of the eluent ions. In some instruments, similar electrolytic strategies are used prior to the analytical column to generate the eluent ions as well. The use of this eluent generation technology leads to less background conductivity and better sensitivity, making it especially useful for the analysis of low levels of ions.
One other way of detecting the ions, and especially anions, was also developed by the chemists at Dow. It involved the use of indirect spectrophotometric detection. In this procedure, an anion such as the phthalate anion is used as the counterion in the mobile phase. The material is usually added as potassium phthalate, and maintained at a concentration of 0.001 M. The phthalate ion absorbs ultraviolet light. The background measurement in the system then consists of a high absorption reading of UV light.

phthalate anion
Suppose we were separating Cl– in a sample containing sodium chloride. Figure 66 shows the end of the column with the chloride ions in the resin, just about to elute in the column. At this point, the concentration of phthalate ion leaving the column is 0.001 M. But what happens when a chloride ion is displaced from the resin to exit the column? This will only happen if a phthalate ion replaces it in the resin. The replacement that occurs will cause the concentration of phthalate ion to drop below 0.001 M.
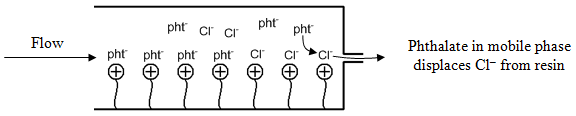
In other words, when the chloride elutes from the column, what you observe in the mobile phase is that the total concentration of anions equals 0.001 M (this has to happen in order to preserve neutrality in the system).
[Cl–] + [pht– ] = 0.001 M
If the concentration of phthalate ion drops proportionally to the concentration of chloride eluting from the column, the UV absorption drops proportionally as well, as shown in Figure 67. The name indirect spectrophotometric detection is aptly chosen since the value of the UV absorption drops as analyte anions elute from the column.
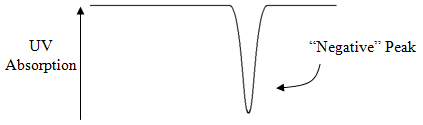
The scientists at Dow Chemical who developed the ion exchange methods for some analyses they needed to perform realized that there were others who would want to be able to measure the same species, and that the device has commercial potential. A company called Dionex, which is still the leading vendor of ion chromatographs, was created to market their invention.