
4.6: Chemiluminescence Imaging
- Page ID
- 76121
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Chemiluminescence imaging combines the sensitive detection of chemiluminescence with the ability to locate and quantify the light emission, but above all it massively provides parallel determinations of the analyte. A digital image is made up of thousands of pixels, each generated by an independent sensor, detecting and measuring the light that falls on it. This enables simultaneous measurement of multiple samples or analytes for high throughput screening. ‘
Chemiluminescence imaging microscopy detects labelled probes more simply and more accurately than does fluorescence. It could become an important tool for rapid, early diagnosis of a wide range of diseases. Whole animal in vivo chemiluminescence imaging makes possible real-time monitoring of pathological and biological phenomena and we may anticipate important advances of great impact in drug discovery, biotechnology and medicine[1].
D6a. Imaging sensors
The last twenty years have witnessed a steady improvement in our ability to form images from analytical signals. Imaging makes use of the high sensitivity and specificity, low background and wide dynamic range of chemiluminescence to quantitate and localize analytes down to the level at which this can be achieved by emission of single photons. Early systems consisted of a low-light vacuum tube device[2] connected to an optical microscope. CCD (charge coupled device) and CMOS (complementary metal oxide semiconductor) image sensors make use of different technologies, both invented in the late 1960s and 1970s, for capturing images digitally. Both types of imager convert light into electrical charge and process it into electronic signals, as in digital cameras. Each has characteristic strengths and weaknesses. CCD technology is in most respects the equal of CMOS. Costs are similar.
a(i). Charge coupled devices
It is central to chemiluminescence imaging that when the spatial distribution of the analyte is critical, a luminograph must be produced to adequately express the data. This can be done by a CCD, which must have a high light-collection efficiency. A CCD converts optical brightness into electrical amplitude signals. CCDs are arrays of semiconductor gates formed on an integrated circuit (IC) or chip. The gates individually collect, temporarily store and transfer charge, which represents a picture element or pixel of an image. When light falls on a CCD sensor, a small electrical charge is generated photoelectrically for each pixel; each charge is converted to voltage and output as an analogue signal, which can be converted to digital information by additional circuitry. All of the pixel can be devoted to light capture. A CCD camera, such as a digital camera includes a CCD imager IC located in the focal plane of the optical system and control circuits mounted on a printed assembly. Image data captured is stored in a storage medium such as a compact flash memory or an IC memory card and can be displayed on a monitor such as a liquid crystal display (LCD). CCDs have traditionally provided the highest image quality (as measured by quantum efficiency and noise). An intensified CCD (ICCD) camera is optically connected to an image intensifier. In an image intensifier, the photons which are coming from the light source fall onto the photocathode, thereby generating photoelectrons. The photoelectrons are accelerated towards a micro-channel plate (MCP) by a voltage applied between photocathode and MCP. The electrons are multiplied by the MCP and accelerated towards a phosphor screen, which converts them back to photons that are guided to the CCD by an optical fibre or lens. ICCD cameras permit high frame rates and real-time visualization, the limitation being the increased noise produced by amplification. Non-intensified slow scan CCD cameras, cooled to reduce thermal noise, permit integration of the signal over a relatively long time and are suitable for steady-state signals.
a(ii). Complementary metal oxide semiconductor (CMOS) chips
CMOS image sensors have emerged as an alternative to CCD sensors. They consist of an integrated circuit (IC) containing an array of pixel sensors, each pixel containing a photodetector, an amplifier and additional circuitry to convert the charge (which represents light intensity) to a voltage. Amplification, noise-correction, and digitization often also occur on-chip. These other functions increase the design complexity and reduce the area available for light capture. Unlike CCD sensors, each pixel is doing its own conversion, so uniformity is lower, which affects image quality. But the chip requires less off-chip circuitry. The phrase "metal oxide semiconductor" implies transistor structure having a metal gate electrode on top of an oxide insulator, which in turn is on top of a semiconductor. CMOS sensors complement every n-type transistor with a p-type, connecting the pairs of gates and drains. Because ideally no current flows except when the inputs to the gates are being switched, this greatly reduces power consumption and avoids overheating, which is a major concern in all ICs. CMOS can potentially be implemented with fewer components, uses less power and provides faster readout than CCDs. CMOS imagers also offer greater integration (more functions on the chip) and smaller size.
It is possible to use a CMOS sensor chip as a microscale contact imager and quantitative photometer for chemiluminescence assays. The applicability has been investigated for chemiluminescence detection of ATP by its reaction with a proprietary reagent in 1 mm diameter wells fabricated on a glass cover-slip placed directly onto the imaging sensor[3]. Ambient light was excluded. For each well, chemiluminescence intensity was averaged over a 1 x 100 pixel region of interest and integrated over a 200 ms exposure. It correlated well with the ATP concentrations over a range of 0.1-1 mmol dm-3. The detectivity (<1 nmol amounts of ATP) is not as fine as can be obtained with a much more expensive CCD camera, but is susceptible to improvement. CMOS chips are suitable for droplet microfluidics or lab-on-a chip devices when the cost of the assay system is a factor that must be optimized, such as in “point-of-need” assays or diagnostics.
a(iii). Photon counting
Cameras suitable for luminescence imaging should be able to form an image at a brightness of 10-6 lux (1 lux = 1/621 W m-3 of 550 nm light). The most sensitive detect single photons with an efficiency of about 20% and have an average noise level of 2 x 10-11 lux = 8 photons s-1 cm-2. Available ultra-low light imaging systems include the imaging photon detector (IPD), used in conjunction with a microscope with high numerical aperture (NA) objective lens. A high NA lens collects more light than a low NA lens. The collected light is focussed onto the IPD. Photons are recorded and stored as a list of time and space coordinates created by the IPD processor. Images can be reconstituted as an array of dots over any desired time interval. This allows for continuous recording over any interval; 1 h at 100 photons per second can be stored in 1 Mb of memory (storage as images requires 0.3 Mb per frame). Chemiluminescence images are based on small numbers of photons, especially when exposure time is brief; whereas a ten minute exposure at 100 photons per second can build an image of 60000 photons, a one second exposure provides only 100 photons, so that the image comprises only 100 dots spread across the area of the image[4] A high resolution (up to 1392 x 1040) single photon counting camera system is suitable for extremely low photon emission applications such as some chemiluminescence applications. The system includes a control unit with data acquisition and image processing software. Frame rates up to 100 Hz can be obtained.
The conceptualization involved in the design of photon counting cameras is well illustrated by DELTA camera; initially designed for astronomy it has advantages for a wide range of high-resolution problems. It is a high sensitivity array detector, which yields the space and time coordinates of photon events at sustained count rates superior to one million per second[5]. It has a flat field, very high resolution (for the prototype: 512 x 591 pixels in space and 2.6 μs in time) and high throughput. Each photon produces an intensified phosphor image which has the same position in a two-dimensional field as the photon. This image is focussed onto three one-dimensional CCDs, which record its position as three coordinates on axes mutually oriented on a plane at angles of 120°. Software converts these to (x, y) coordinates on orthogonal axes and a clock signal adds the time (t) of the event to produce (x, y, t) coordinates, which are listed and stored; artefacts due to excessive tolerance or to simultaneous photon events are also removed from the data
a(iv). Chemiluminescence imaging systems
A high resolution CCD camera (up to about 6.0 Mpixel), cooled to about -70ºC, gives the best quality images, better accuracy, longer exposure times (up to 24 hours), minimal dark noise and enhanced stability. There should be a light-tight dark chamber and a height adjustable sample platform with a numeric counter for exact positioning in specific, repeatable positions. The alternative is an advanced motorised robotic camera which is driven up and down allowing placement very close to the sample, outperforming a standard zoom lens, having a wider field of view, easier and faster operation, and better sensitivity. These benefits are especially useful for faint samples. The image acquisition and analysis software provides comprehensive tools for simple image capture and analysis of gels, plates and membranes as well as colony counting. Images can be enhanced, user preferences defined, reports generated and data exported. Some systems include an overhead white light so that chemiluminescence can be overlaid onto a reflected light image (the so-called “live image”) and combine facilities for fluorescence, chemiluminescence and colorimetric applications.
Not all chemiluminescent reactions are suitable for imaging; the main requirement, especially for imaging microscopy, is micrometre scale localization[1]. Excited species of short half-life are suitable and conditions (especially reactant concentrations) can be optimized to minimize the diffusion of excited products. Glow-type kinetics, arising from the attainment of a steady state, facilitates measurement procedures. Enzyme labels are widely used.
D6b. High throughput screening (HTS)
Imaging is very suitable for high-throughput screening. Examples of assays with very high sample throughput include the determination of bioavailable mercury which has been determined in urine using E. coli expression of luciferase under the control of a mercury-inducible promoter. Throughput is more than 5000 samples per hour and the limit of detection is 10-13 mol dm-3. Acetylcholinesterase inhibitors can be assayed using acetylcholinesterase, choline oxidase and horseradish peroxidase (HRP). Kinetic analysis of luminol chemiluminescence is carried out with a throughput of 180-360 samples per hour.
b(i). Miniarrays
Imaging with flat field correction lenses can be used to read microtitre plates (up to 4 of 384 wells) faster than by a luminometer, but the latter is, however, more sensitive and has the ability to measure fast, flash-type reactions. Miniarrays of antibody or gene probes can be spotted onto 96-well microtitre plates and assayed with an enzyme-labelled detection reagent and a chemiluminescence substrate. The whole plate is imaged with a CCD camera to measure the light emission from each well. This principle has been applied to a sandwich-type enzyme-linked immunosorbent assay (ELISA) for cytokines and a hybidization-based mRNA assay with up to 16 spots in each well; a 16 x 96 array contains 1536 dots, making possible high-throughput multianalyte assays, using standard plates and their associated sample handling devices.
Oligonucleotide probes specific for human papilloma virus (HPV) genotypes have been used for a multianalyte chemiluminescence imaging assay for the simultaneous determination of up to 7 HPV DNAs. Amplification by the polymerase chain reaction(PCR) in the presence of a digoxigenin-labelled nucleotide (dUTP) is followed by an ELISA using a novel polystyrene microtitre plate having an array of 24 main wells (containing digoxigenin-labelled PCR product) each divided into 7 subwells (containing the immobilized probes). The digoxigenin label was subsequently detected by peroxidase-labelled antibody and a chemiluminescent substrate. Imaging was performed using an ultrasensitive CCD camera[6]. Results were comparable with conventional colorimetric PCR-ELISA.
b(ii). Microarrays
Thousands of simultaneous determinations can be made by high-resolution imaging of chemiluminescence at array densities of up to hundreds of spots per square centimetre. Array-based gene expression analysis is a good example. Protein analysis based on antigen-antibody or ligand-receptor interactions is increasingly used in clinical and research work and in drug discovery. As well as their use for protein expression profiling, there are high-throughput protein microarrays that detect up to 35 cytokines. Specific antibodies are spotted onto membranes, which are incubated with the samples and captured analytes are detected by enhanced chemiluminescence with HRP-labelled antibodies and an HRP-substrate. A protein chip for parallel ELISAs of tumour marker allows the discovery of patterns that can increase the sensitivity and specificity of the diagnosis[7]. Immobilized on the chip were 12 monoclonal antibodies against different tumour markers captured by incubating the chip with serum samples. An HRP-conjugated second antibody was used for detection by chemiluminescence imaging. The chip has been successfully applied both for cancer diagnosis and for screening asymptomatic populations at high risk.
b(iii). Small scale analytical devices
Small scale analytical devices use extremely small sample volumes and so need very sensitive detection techniques; chemiluminescence imaging has the high resolution and high sensitivity necessary. Assays include the ELISA determinations of the herbicide 2,4-D in multiple samples using gold-coated surfaces or glass capillaries and of up to ten antibiotics in milk in five minutes: the sample is incubated with mixed monoclonal antibodies followed by detection with an HRP-labelled second antibody and a suitable chemiluminescent substrate. Multiple hybridizations can be performed in a three-dimensional chip incorporating an array of vertical glass channels. Specific gene probes are immobilized on the inner walls of the channels. This strengthens the signal by providing a larger area for probe-immobilization than is available in a two-dimensional microarray. The sample flows through the channels and the analyte is detected by an enzyme-labelled antibody followed by a chemiluminescent substrate. Lateral diffusion of the emitting species is prevented by the walls of the microchannel; this improves resolution of the image. Chemiluminescence imaging of miniaturized analytical devices is also useful for multiplexing (simultaneous quantitation of different analytes or on different samples) by integrating the chemiluminescence over a different target area for each analyte or sample.
b(iv). Documentation of gels and membranes
Chemiluminescence imaging detection with CCD cameras can be used for reactions that take place on gels and membranes. This allows intensity measurement over a wide dynamic range and software exists to compute the total emission from particular zones of the image for analytical purposes. Images can be stored on discs or printed out.
Electrophoresis is the movement, and hence the separation, of charged molecules in an electrical field; electrophoresis on polyacrylamide gel (PAGE) is particularly good for separation of molecules at low concentrations. Separated molecules can be transferred onto a nitrocellulose membrane by electroblotting - electrophoresis in a direction at right angles to the gel surface; this is also called western blotting and it is used to detect specific proteins in a sample of tissue homogenate or extract. By similar procedures, Southern blotting identifies particular sequences of DNA within a complex mixture and northern blotting locates RNA. In western blotting, electroblotting is followed by immunostaining, in which particular proteins are identified by labelled antibodies. DNA and RNA can be similarly identified by hybridization with labelled probes.
Dot blot is an immunological technique to detect with antibodies specific proteins in mixtures or in samples such as tissue lysates. It is based on western blotting but there is no separation of the protein on SDS-PAGE. One such assay is the detection of B19 parvovirus. After spotting samples onto a membrane, hybridization with digoxigenin-conjugated DNA probes and treatment with HRP- or AP-labelled anti-digoxigenin antibodies, chemiluminescence imaging gives a limit of detection ten times better than using colorimetry.
Cytochromes can be separated by PAGE with sodium dodecyl sulfate (SDS-PAGE) and transferred to a nitrocellulose membrane; cytochrome c (which contains a catalytic haeme group) is detected by peroxidase-luminol chemiluminescence[8]. CCD imaging results in detection 50 times more sensitive than the 3,3',5,5'-tetramethylbenzidine staining method. A sample of less than 1 ml of a bacterial culture is needed. A similar assay, based on luminol/hydrogen peroxide chemiluminescence with ammonium persulfate enhancement, detects haptoglobin phenotyping after PAGE on a 15 μL sample[9]. Other iron-containing proteins, such as catalase and ferritin, can also be detected. The proposed detection is very fast, compared to traditional staining methods (minutes versus hours).
D6c. Molecularly Imprinted Polymers (MIPs)
MIPs have artificial recognition sites with shapes, sizes and functionalities complementary to the analyte, which is thus selected in preference to other closely related structures. They are cheaper and more robust than antibodies, enzymes and biological receptors and can serve when these biomolecules are not available. The recognition sites are fabricated around a suitable template, preferably the analyte itself, which is extracted after polymerization. Generally, when a template molecule and a functional monomer are mixed in an organic solvent a complex is formed between the template and the monomer through polar interactions. Polymerization with a cross-linker fixes the positions of the polar groups. Removal of the template with a suitable solvent leaves specific recognition sites. The functional monomers are chosen to promote hydrogen bonding with the template to obtain good selectivity and reversibility. Optimum binding occurs when the MIP is exposed to the same conditions as those used in polymerization, because it depends on the shape of the imprinted cavity and on the spatial positioning of the coordinated functional groups. Both of these depend on the conditions and are affected by swelling of the polymer, which can be exploited to achieve fast and controllable release of adsorbed molecules prior to detection.
c(i). Chiral recognition of dansyl-phenylalanine
Molecular imprinting of polymers has been linked with chemiluminescence imaging detection to achieve chiral recognition of dansyl derivatives of phenylalanine (Phe)[10]. The MIP microspheres were synthesized using precipitation polymerization (which produces uniform microspheres) with dansyl-L-Phe as template and the microspheres were immobilized on microtitre plates (96 wells) using poly(vinyl alcohol) (PVA) as glue. The analyte was selectively adsorbed onto the MIP microspheres. After washing, the bound fraction was quantified using peroxyoxalate chemiluminescence (POCL) analysis, a general method for all fluorescent and fluorescence-labelled analytes, which has a greater quantum yield than most other chemiluminescence systems. In the presence of dansyl-Phe, bis(2,4,6-trichlorophenyl)oxalate reacted with hydrogen peroxide (H2O2) with chemiluminescence emission. The signal was detected and quantified with a highly sensitive cooled CCD. The intensity of the image of each well of the plate was determined using software to sum the intensities of all the pixels making up the spot. Chemiluminescence intensity increases with the proportion of the L-enantiomer in the sample. Chiral composition can thus be determined by comparison of the intensity for the mixture and for pure D- and L- enantiomers at the same concentration. The results show that MIP-based chemiluminescence imaging is useful for quick chiral recognition and, because the method can perform many independent measurements simultaneously in 30 min, high-throughput screening is possible.
c(ii). High throughput detection of dipyridamole
A simple, sensitive and specific method has been developed for high throughput detection of the vasodilator dipyridamole[11]. The proposed method is based on a chemiluminescence imaging assay with MIP recognition providing selectivity.
Molecularly imprinted microspheres were prepared using precipitation polymerization with methacrylic acid (MAA) as functional monomer, trimethylolpropane trimethacrylate (TRIM) as the crosslinker and dipyridamole as the template. Non-imprinted polymer (NIP) was prepared without template to use as a control. The microspheres were coated in 96-well microtitre plates using 0.1% PVA as glue. After incubation with the sample, the amount of polymer-bound dipyridamole was determined by POCL. The emitted light was measured with a cooled high-resolution CCD camera. The intensity of the image of each well was determined as in subsection c(i).
Under the optimum conditions, there is a linear relationship between relative chemiluminescence intensity and concentration of dipyridamole ranging from 0.02 to 10 μg ml-1. The detection limit is 0.006 μg ml-1. The method was validated by measuring dipyridamole concentrations in spiked urine samples. High tolerance for a number of normal constituents of urine was demonstrated to be much greater in the presence of MIP rather than NIP. MIP-based chemiluminescence imaging exhibits high selectivity and sensitivity to dipyridamole, combined with high sample throughput and economy (50 μl/well)[12].
D6d. Spatial distribution of targets
Target molecules of chemiluminescence imaging include antigens, DNA sequences, enzymes and metabolites. Chemical processes in cells, tissues or whole animals may also be targeted. Methods used include imaging microscopy, immunohistochemistry (IHC), in situ hybridization (ISH); other chemical or enzymatic reactions may also be used. The chemiluminescence image is overlaid onto the visible light image and processed by background subtraction, contrast enhancement, pseudocolour and quantitation over defined areas; absolute quantitation needs reproducible conditions, a calibration system and appropriate sample properties.
d(i). Imaging microscopy
Chemiluminescence imaging microscopy uses ordinary microscopes with optimized light collection. Light loss is minimized by having a simple lens coupling system; coverslips are dispensed with. The microscope, or at least the sample, is contained in a dark box to exclude ambient light and has a motorized micrometric stage to permit automatic adjustment. The sample is incubated with the chemiluminescence reagent until a steady-state emission is obtained. The objective lens has the highest numerical aperture (NA) compatible with acceptable focal aberration and depth of field. Dry, rather than oil-immersion, objectives are used and give adequate magnification and spatial resolution for the localization of analytes in single cells or tissue sections[13]; the detection limit for HRP is about 500 molecules/μm2.[14]
Chemiluminescence microscopy has become a standard tool in biomedical research. Photon detectors have been attached to microscopes and allow imaging of chemiluminescent probes and reporter genes in cells and tissues. Photon counting techniques allow days of continuous imaging without creating oversized files. Fluorescence imaging, however, gives better spatial resolution than chemiluminescence imaging and makes multiple determinations easier.
d(ii). Calcium imaging with chemiluminescence microscopy
Calcium can be determined in cytosol and in organelles by using the photoprotein aequorin, an intracellular calcium indicator extracted from the jellyfish Aequorea victoria. Natural aequorin consists of a polypeptide, apo-aequorin, covalently bound to a hydrophobic prosthetic group, coelenterazine. The principle of imaging free cytosolic calcium with aequorins[3] is the conformational change of aequorin molecules on calcium binding, causing coelenterazine to be oxidized to coelenteramide with production of carbon dioxide and emission of blue light (466 nm). Aequorin cannot penetrate the plasma membrane of the cell. Microinjection is the method of choice for determining cytosolic calcium in large cells. For small cells, cloning and transfection of the cDNA of apo-aequorin makes microinjection unnecessary, greatly simplifying calcium recording. Genetically expressed apo-aequorin contains no coelenterazine and so does not emit light. It is reconstituted as aequorin by soaking the specimens with coelenterazine. Apo-aequorin can be targeted to specific organelles by incorporating signal translocation sequences in the polypeptide chain.
Aequorin is sensitive and specific, though single cells, containing a low concentration, give feeble chemiluminescence. Intensity is proportional to cell volume and therefore to the cube of the diameter. Small cells present problems because the amount of aequorin is low. In a cell of 10 μm diameter, the resting calcium concentration leads to emission of less than one photon per hour – so fluorescence must be used instead. But elevated calcium concentrations or large numbers of cells can be imaged by chemiluminescence using photon-counting cameras. Natural aequorin accurately measures Ca2+ concentrations in the range 0.5 to 10 μmol dm-3, which is suitable for transient changes but for higher concentrations, a mutant form has been constructed which, by raising its dissociation constant and thus lowering its affinity for calcium, extends the working range up to 100 to 1000 μmol dm-3. It has the advantage over calcium-specific fluorescent probes of permitting real-time measurements over a long period; this is possible as there is no disturbance of the intracellular environment (including Ca2+ buffering capacity) because of the low aequorin concentration (about 5 nmol dm-3) but it has poorer resolution and it is used up rapidly by high calcium concentrations. Aequorin chemiluminescence, however, has an excellent signal to noise ratio and extremely low background noise.
Chemiluminescence calcium imaging using aequorin is the method of choice for exploratory studies, since it is extremely sensitive, can detect a broad range of calcium concentrations. The kinetic order with respect to calcium concentration of the chemiluminescence reaction is 2.1 or higher, which gives inherent contrast enhancement. Unlike fluorescence, it does not require the analyst to make preliminary predictions or assumptions which exclude calcium signals outside the expected range. But it cannot match the high spatial resolution of fluorescence methods. In addition, chemiluminescence microscopy uses a large depth of field and optical sections are not yet possible.
d(iii). Aequorin associated with green fluorescent protein (GFP)
In Aequorea victoria, the chemiluminescent calcium-binding protein, aequorin, is associated with GFP. Calcium-sensitive bioluminescent reporter genes have been constructed that fuse GFP and aequorin to increase the quantum yield of calcium-induced bioluminescence[15]. Co-expression of GFP with free aequorin does not have the same effect. The constructs were varied by including different lengths of peptide spacer between the GFP and the aequorin; much more light was emitted in all cases and the constructs were much more stable in cytosol and more sensitive to calcium than recombinant apo-aequorin alone.
Resonance (non-radiative) energy transfer to the GFP chromophore from the excited oxidation product of coelenterazine depends on their relative positions. The peptide spacer is therefore flexible and of variable length. The green:blue ratio (500 nm:450 nm) of the light emitted by different constructs was measured 48 h after the introduction of the reporter genes into the cells (transfection). The green:blue ratio was increased by the covalent attachment of GFP to aequorin and further increased when the linker was added; as the linker was made longer, the wavelength of maximum emission increased and the bandwidth of the spectrum decreased. The efficiency of intramolecular energy transfer is enhanced to a level comparable to that achieved by resonance energy transfer in vivo due to the more favourable configuration made possible by the linker.
Using GFP-aequorin fusions it is possible to detect physiological calcium signals in single cells. Transfection of the cells is followed by aequorin reconstitution with coelenterazine. The result is calcium-induced photon emission detectable with a cooled, ICCD camera, using an integration time of only one second. Cytoplasmic aequorin had previously detected Ca2+ activities only by the use of a photomultiplier, which is more sensitive but lacks any spatial resolution, or by using targeted fluorescence probes, which give a quicker response. The use of the transgenes in which aequorin reports Ca2+ activity while GFP enhances bioluminescence could lead to real time imaging of calcium oscillations in integrated neural circuits in whole animals as well as in specific subcellular compartments. Aequorin and GFP-enhancement probes along with synthetic fluorescent dyes can be targeted to the endoplasmic reticulum (ER)[16], a membrane network within the cytoplasm of cells involved in the synthesis, modification, and transport of cellular materials; this has enabled the role of ER to be clarified.
D6e. Enzyme and metabolite mapping
If the sample’s enzyme activity has been preserved and if the sample is in such a condition that the substrate has access to the active site, enzyme activity can be localized by chemiluminescence imaging. The best spatial resolution is obtained by applying a chemiluminescent substrate directly to an enzyme, e.g., alkaline phosphatise can be detected by dioxetane phosphate. Enzyme reactions coupled with chemiluminescence can be used for metabolite mapping but give lower resolution. Metabolites can be determined in shock frozen tissue biopsies at femtomole levels and with micrometre resolution. The tissue is frozen as soon as possible to stop enzyme activity and fix the metabolites. The specimen is then placed on a temperature-controlled microscope stage and the chemiluminescence reagent is added. Emission intensity, recorded as soon as the temperature rises sufficiently, is converted into metabolite concentrations.
e(i). Luciferase-based detection of energy metabolites
Measurement[17] of the spatial distribution of metabolites, such as ATP, glucose, and lactate, in rapidly frozen tissue is based on enzymatic reactions that link the metabolites to luciferase with subsequent light emission. Using an array, cryosections are brought into contact with the enzymes in a reproducible way inducing emission of light from the section in proportion to the metabolite concentration, with high spatial resolution. There is a close correlation between the distribution of ATP and cell viability; there are also distribution differences between tumours and normal tissue. ATP, glucose, glycogen and lactate have been determined at microscopic levels and at high spatial resolution in arterial wall cryosections using luciferase-based chemiluminescence imaging[18], which is a powerful tool to measure energy metabolites. It has been used to quantify local metabolite concentrations in artery rings. Distributions of energy metabolites are heterogeneous under hypoxic in vitro conditions. Diffusion distances for oxygen and nutrients can be long and might make vessels prone to develop local deficiencies in energy metabolism that could contribute to atherogenesis.
e(ii). Expression of luciferases in living cells and organisms
Luciferases are enzymes that emit light in the presence of oxygen and a luciferin (ADD LINK). They have been used for real-time, low-light imaging of gene expression; coding sequences have been detected by luciferase-labelled gene probes[19] . These labels include bacterial lux and eukaryotic luciferase luc and ruc genes. Different luciferases differ in the stability/variability of the emitted signal. Luciferases have served as reporters in a number of promoter search and targeted gene expression experiments. Photon-counting CCD imaging of luciferase has been used, for example, to show promoter activity in single pancreatic islet β-cells and regulation of human immunodeficiency virus (HIV) and cytomegalovirus. Luciferase imaging has also been used to trace bacterial and viral infection in vivo and to visualize the proliferation of tumour cells in animal models. Infected cells are readily detectable at an incidence of one in a million cells. Single bacterial cells, whether transformed or naturally luminescent, have also been imaged and variation in expression over time, due to fluctuations in metabolic activity, has been demonstrated. Low-light CCD imaging is in itself a non-invasive technique that is useful for observing (see subsection b(iv). Documentation of gels and membranes ADD LINK) intracellular gene expression and small-scale assays such as in situ hybridization (ISH) as well as for immunoassays, gels and blots [ADD LINK], DNA probes and in vivo imaging (see section D6h, ADD LINK). Slow-scan liquid nitrogen cooled CCD cameras are preferable for high resolution imaging with long exposures, but photon-counting CCD cameras are better for shorter exposure times. Flashing at frequencies greater than 1 Hz can be detected by ICCD cameras.
e(iii). Other applications of bioluminescence imaging
Bioluminescence imaging has been applied in experimental biomedical research, e.g., development of necrosis, and in other areas of biology[20]. It has also been used particularly on tumour biopsies in clinical oncology. In combination with immunohistochemistry,autoradiography or in situ hybridization it can be particularly powerful. It has been shown for squamous cell carcinomas that accumulation of lactate in the primary lesions is associated with a high risk of metastasis. In this way, metabolic mapping indicates the degree of malignancy and the prognosis of tumours; it has stimulated a number of fundamental investigations.
e(iv). Other methods of determination of metabolites
There are numerous other examples of methods to determine metabolites in living cells and tissues, including real-time imaging of metabolite production. Endogenous acetylcholinesterase (ACE) activity has been detected in rat coronal brain slices using coupled reactions with choline oxidase and horseradish peroxidase[21]. The reagent is optimized to minimize diffusion of emitting species, giving sharp localization and a very low background. This imaging assay is more predictive than in vitro systems; and can be used to determine pathophysiological changes in ACE distribution or the effect of in vivo ACE inhibitors, which could be useful for screening candidate drugs.
Nitric oxide (NO) released from cell cultures and living tissue has been visualized by a reaction with luminol and hydrogen peroxide to yield photons which were counted using a microscope coupled to a photon counting camera, giving new insight into release time course and diffusion profile[22]. The method allowed integration times in the order of minutes to improve signal-to-noise ratio. However, the high sensitivity of this method also makes it possible to generate an image in seconds, allowing the production of real time moving pictures. This method has demonstrated potential for real time imaging of NO formation, with high temporal and spatial resolution. There was little earlier knowledge of this phenomenon due to the short half-life of NO.
D6f. In situ hybridization (ISH) and immunohistochemistry (IHC)
ISH and IHC are techniques that localize analytes in a wide range of suitable specimens such as cellular smears, or frozen or paraffin-embedded sections. Chemiluminescence detection does not require any special specimen preparation, but an accurately controlled section thickness of from 3 to 5 μm is necessary for reproducibility. Incorporating chemiluminescence detection (CL-IHC and CL-ISH) increases sensitivity compared with colorimetry or fluorometry. This adds reliable and accurate quantitative evaluation of spatial distribution to the specificity of the probe. The “theoretical” limit of detection of the enzyme label by chemiluminescence is 10-21 to 10-18 mol; as a detector for ISH,chemiluminescence is almost as sensitive as 35S autoradiography giving a nontoxic alternative to the use of radioactivity[23].
Localization within cells of nucleic acid sequences, e.g., the sites of genes in chromosomes, can be achieved by hybridization to complementary nucleic acid probes. The two general types of in situ hybridization involve nuclear DNA and cellular RNA respectively; they are conceptually similar but differ in practical detail. The technique is usually performed on specimens prepared for light microscopy. It is claimed that little or no microscopic training is necessary to evaluate the chemiluminescence images.
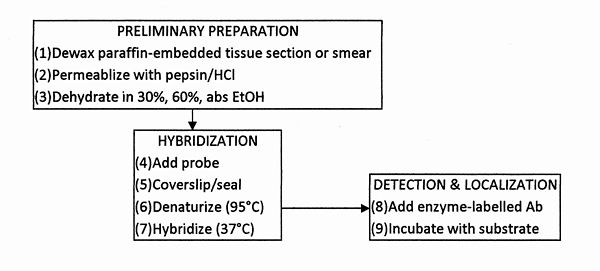
Fig. D6.1 – Flow chart of operations for the performance of in situ hybridization. (All incubations are at room temperature).
f(i). CL-ISH assay of human papilloma virus (HPV)
ISH involves nucleic acid probe hybridization with DNA or RNA, endogenous to the specimen or exogenous (viral/bacterial). The procedure is summarized in Fig. D6.1. Sensitivity is increased by indirect labelling, in which the label binds with a biospecific chemiluminescence reagent, e.g., biotin binds to streptavidin; fluorescein or digoxigenin bind to their respective antibodies, the chemiluminescence reagent having a covalently-bound signalling group (usually AP or HRP). ISH of HPV can be imaged using a digoxigenin-labelled gene probe followed by an HRP-labelled anti-digoxigenin antibody and a chemiluminescence reagent[24] . To localize the virus, the chemiluminescence image (with intensities represented by pseudocolours) is overlayed onto a transmitted light image. ISH has also been performed on three human carcinoma cell lines and 40 biopsy specimens of human cervical neoplastic and preneoplastic lesions by using biotin-labelled complementary DNA probes of HPV, detected by HRP-labelled secondary antibodies; the chemiluminescence was detected by an ICCD camera[15]. After only 10 min of photon accumulation, on cell line smears as well as on serial tissue sections, chemiluminescence gave comparable results to those obtained by a 3-week exposure for 35S-autoradiography.
CL-ISH is quantitative because chemiluminescence is proportional to the enzyme activity of the label, and to the number of gene copies per cell. In a separate study of cytomegalovirus, chemiluminescence was proportional to the number of cells infected (following virus replication).
f(ii). CL-ISH of cytomegalovirus
An early ISH assay of cytomegalovirus DNA in infected human fibroblasts[13] used dioxigenin-labelled probes and AP-labelled anti-digoxigenin antibody. Employing a low-light imaging luminograph 400 amol of AP were detected using 1,2-dioxetanes . Chemiluminescence was intense and stable, making possible quantitation within single cells, with a spatial resolution of 1 μm and very low background. Multiplexed CL-ISH assays have been developed in which probes with different enzyme labels detect different targets. One example of such techniques localizes the DNA of herpes simplex and cytomegalovirus in the same specimen using the following protocol. The faster HRP/luminol is added to the specimen and chemiluminescence is imaged, the specimen is given a short wash, then AP/dioxetane is added, and a second chemiluminescence image is recorded. A longer wash is needed if AP is added first.
f(iii). CL-ISH of parvovirus B19 nucleic acids in single infected cells
Human parvovirus B19 is responsible for wide range of diseases. CL-ISH gives high resolution, providing precise localization and quantitative detection of the viral nucleic acids in single cells in cultures at different times after infection, giving an objective evaluation of the infection process with higher sensitivity than colorimetric ISH detection assessed by panels of observers. The improved sensitivity of CL-ISH detects more positive cells per sample, making possible earlier diagnosis.
A peptide nucleic acid (PNA) has been developed which has improved specificity and faster, stronger binding than other DNA probes. The assay is based on the use of a biotin-labelled PNA probe which is detected by a streptavidin-linked alkaline phosphatase (AP), using the well-known biotin-streptavidin affinity:
PNA–biotin + streptavidin–AP → PNA–biotin–streptavidin–AP
adamantyl 1,2-dioxetane phosphate + AP → excited fragmentation products → light
The chemiluminescence signal which arises was quantified and imaged with an ultrasensitive nitrogen-cooled CCD camera connected to an epifluorescence microscope with high-transmission optics and modified for acquisition of chemiluminescence. A threshold signal (representing non-specific binding of the probe and endogenous alkaline phosphatase activity) was established using mock-infected cells as negative controls. Following a B19 virus infectious cycle the percentage of infected cells, which reached its maximum at 24 h after infection, could be accurately monitored. The advantages of chemiluminescence detection (high detectability and wide linear range) allow the quantitative analysis of viral nucleic acids in infected single cells, showing a continuous increase with time after infection. Such investigations could be powerful tools for the assessment and diagnosis of viral infections and for measuring the virus load of infected cells[25].
f(iv). IHC with chemiluminescence detection (CL-IHC)
IHC involves the use of antibodies that bind to endogenous, viral or bacterial antigens (a protein usually) with subsequent detection by enzyme-conjugated antibodies. CL-IHC detects epithelium in thyroid tissue by HRP-labelled antibodies and luminol/H2O2, with adequate resolution and greater sensitivity than colorimetry or fluorescence. CL-IHC can also with advantage be applied to Interleukin 8 (IL-8) localization in gastric biopsy specimens infected by Helicobacter pylori, an organism asssociate with gastric ulcers. It shows with greater sensitivity than other detection systems the variability of the IL-8 concentration in the mucosa and the foci of high concentration in the epithelial cells.
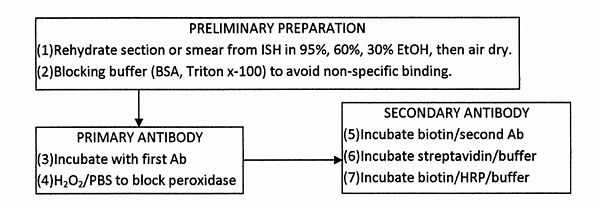
Fig. D6.2 – Flow chart of operations for the performance of immunohistochemistry. (All incubations are at room temperature).
f(v). HPV and p16(INK4A) marker in cervical cancer
Cervical cancers (cervical intraepithelial neoplasms, CIN) are classified into low- (CIN1) or high-grade (CIN2 or CIN3) in order to predict the risk of progression of early lesions and enable decisions to be made concerning surgical intervention. Judgments based on histology are imprecise in that different observers assign different grades to the same biopsy specimen. One way of overcoming this difficulty is to redefine the diagnostic criteria in terms of analytical chemistry.
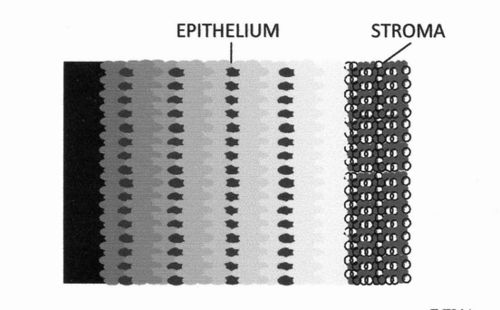
Diagrammatic representation of the localization of p16INK4A by CL-IHC in a biopsy section of a cervical cancer. Paler shades show increased chemiluminescence emission. (An actual chemiluminescence image is shown in Fig. 1 in reference 27, on which this diagram is based.)
An immunohistochemical assay (see Fig. D6.2) with chemiluminescence detection (CL-IHC) has been used to quantitatively evaluate the overexpression of the protein p16INK4A and its localization in the epithelium of samples from cervical cancers and from non-cancerous cervical lesions. Fig. D6.3 shows that chemiluminescence (and hence p16INK4A protein content) generally increases from left to right. High-grade lesions give generally more intense chemiluminescence signals in the epithelium than low-grade and show a different distribution of p16INK4A protein. From the intensity of the chemiluminescence signal and the percentage of the epithelium involved in the overexpression of p16INK4A an expression score was obtained which discriminated well among different lesions. A cut-off value was determined to distinguish between low and high grades. The differences between the average scores of different CIN grades were statistically significant[26].

Diagrammatic representation of the co-localization of p16INK4A and HPV DNA in a tissue section from a cervical biopsy. (A) transmitted light microphotographic image, (B) CL-IHC image, the lighter tones showing p16INK4A, (C) CL-ISH image, the lighter tones showing HPV DNA, (D) CL-ISH image pseudocoloured in blue, yellow and red to indicate increasing chemiluminescence intensity. (B), (C) and (D) show images overlaid onto a transmitted light image to show the localization of the signal. (Actual chemiluminescence images are shown in Figs. 3 and 4 in reference 27, on which this diagram is based.)
The determination of p16INK4A overexpression by CL-IHC used AP as the enzyme label and then, after washing in buffer, HPV DNA was determined by CL-ISH[27] (see Fig. D6.1 and section f(i) ADD LINKS) using HRP as label to avoid interference between the two assays[28]. To circumvent the non-equivalence of consecutive tissue sections, the two assays were carried out on the same sample. The assays cannot be carried out in the reverse order as the high-temperature step in ISH denatures the p16 protein to be determined by IHC. The high detectability of chemiluminescence gives improved discrimination between lesions as non-cancerous, CIN1 or high-grade CIN. This could become an objective and accurate diagnostic test.
f(vi). Mucosal human papilloma virus in malignant melanomas
High-risk (HR) mucosal human papilloma virus (HPV) is strongly associated with cancer. It has been found in primary melanoma and in pigmented skin blemishes (birthmarks, moles) but has rarely been reported in normal skin, which is instead commonly infected with other relatively harmless strains of HPV. HPV DNA in skin cancer has been detected by polymerase chain reaction (PCR). In order to understand the relationship between HPVs and primary melanoma, it is necessary to know whether the presence of HPV is localized in cancer cells rather than in normal skin cells present in the tumour biopsy, what proportion of the cells harbours the virus or whether it can be due to contamination of the tumour surface by viruses from healthy skin. Because PCR methods measure only total DNA they are not suitable to ascertain this.

Schematic representation of a tissue section that has undergone the combined procedures of FL-ISH for HPV DNA and CL-IHC for tumour marker HMB-45. The large coloured dots represent cells. Red pseudocolour was assigned to chemiluminescence signals and yellow to fluorescence signals, different signal intensities represented by different shades. Colocalization of HPV and HMB-45 is represented by the combined pseudocolour, orange.
To localize HR-HPV a rapid, specific and very sensitive method has been developed that combines an enzyme-amplified fluorescence in situ hybridization (FL-ISH, see Fig. D6.1 ADD LINK) for the detection of HPV nucleic acids (types 16 and 18, which are the types most likely to cause cancer) with a chemiluminescence immunohistochemistry (CL-IHC, see Fig. D6.2 ADD LINK) method for the detection sequentially in the same section of the tumoural melanocytic marker HMB-45. It is necessary to use the same section because the melanoma cells are distributed heterogeneously in the specimens. HMB-45 determination is an indicator of melanoma cell differentiation and is widely used in diagnostic pathology. Digital images of FL-ISH and CL-IHC were separately recorded, assigned different pseudocolours (see Fig. D6.5) and merged using specific software for image analysis. The results demonstrated a sharp colocalization (to an extent of about 70% of the total luminescent area of the specimen) of HPV nucleic acids and the melanoma marker in the same biopsy sections. In smaller areas, HPV was detected without HMB-45 (9.5% of total) or HMB-45 without HPV (20.5%). This demonstrates that viral nucleic acids were specifically present in melanoma cells and supports a possible active role of HPV in malignant melanoma[29].
D6g. Chemiluminescence imaging of fluorescence reporters
Fluorescence detection remains of great value for in situ hybridization and in immunohistochemistry, particularly because of its greater precision of spatial localization compared with chemiluminescence. Chemiluminescence can, however, be harnessed as a means of exciting fluorescent probes and labels alternative to photo-excitation. Two examples of the principle are considered in this section.
g(i). Peroxy-oxalate chemiluminescence
Using imaging chip-based devices, detection of aqueous peroxyoxalate chemiluminescence (POCL) from oxamide I in aqueous environments has been reported[30] for fluorescence-labelled analytes and proved to be at least as sensitive as that using direct fluorescence detection requiring a light source for excitation. Using a CCD camera to record the chemiluminescence intensity from a 1000-fold range of analyte concentrations, POCL detection sensitivity of fluorescence-labelled immunoglobulins on a nitrocellulose membrane was investigated. Aqueous POCL of Staphylococcus aureus enterotoxin B (SEB) and its antibody were also used to demonstrate immuno- and affinity-detection using a CCD camera. SEB was detected by an immune sandwich assay in which SEB was captured by sheep polyclonal antibody spotted onto a nitrocellulose membrane and subsequently captured mouse monoclonal antibody, which was detected by fluorescence-labelled anti-mouse antibody. Affinity detection of biotin-labelled anti-SEB antibody used fluorescence-labelled streptavidin.
Simultaneous detection by POCL of bovine serum albumin labelled with two different fluorescent labels has been demonstrated, using contact imaging with a CMOS colour imaging chip (ADD LINK). The proteins were spotted onto a membrane disc fixed to a cover slip which was placed on the sensing surface of the chip. They were visible on 8 s exposure as red and green spots respectively; a mixture of the labelled samples emitted yellow light. This procedure might be applicable to reading microarrays.
g(ii). Bioluminescence resonance energy transfer (BRET)
A self-illuminating fluorescence reporter, comprising a dye conjugated to AP, has been demonstrated “in principle” for imaging detection using a CCD camera or a CMOS colour chip, making possible the imaging of fluorescent signals without the need for an external light source or sophisticated optics[31]. It is based on bioluminescence resonance energy transfer (BRET), an example of which, already cited, is the use of GFP to enhance the light emission from the photoprotein aequorin (see section d(iii) ADD LINK). The efficiency of BRET is increased by minimizing the distance between the bioluminescent energy donor and the fluorescent acceptor and is also found to depend on the ratio of AP to fluorophore in the conjugate, on the fluorescent dye used and on the chemiluminescent substrate. Chemiluminescence detection is low-cost, suitable for low concentrations and portable but diffusion of the luminescent products leads to poor spatial resolution. BRET is a potential solution to this problem, but it has not yet been applied to a real analytical problem.
In the demonstration, antibody, immobilized on the CMOS surface, captured a biotin-labelled target molecule that was then bound to the streptavidin-labelled AP-dye conjugate. The AP was used to generate light and the captured array images were viewed on a computer monitor. Images were also obtained by using a CCD camera. The chemiluminescent substrate for AP emitted at 450 nm; the energy from this emission was transferred to the fluorescent dye. This resulted in a second light emission with a longer wavelength (580 nm), which was localized at the position of target molecules, avoiding the problem of diffusion of the chemiluminescent product. In this way, image spatial resolution was greatly improved compared with conventional chemiluminescence detection. The shorter wavelength first emission that escaped absorption by the dye was removed by a high pass filter.
D6h. Whole-organ and whole-organism imaging
The use of cameras remote from the site of light emission makes it possible to image events occurring in the interior of organs and organisms. This technique can be applied to the study of a wide range of phenomena such as tumour growth, metastasis and drug efficacy, assessed by injecting and imaging recombinant light-emitting tumour cells[32], which can be used as probes for tumour location. Another application of molecular imaging techniques is the non-invasive monitoring of transplanted embryonic cardiomyoblasts expressing firefly luciferase (Fluc) reporter gene[33]. The movement in the rat gut of bioluminesent E. coli (expressing luciferase and the enzyme necessary for substrate synthesis) has also been imaged[34].

Diagrammatic representation of Renilla luciferase expression in a transgenic tobacco leaf, imaged against a dark background.. Light emission is indicated by white or grey areas. (The diagram is based on the image reproduced in figure 1 of reference 34)
Luciferase enzymes to label cells, pathogens, and genes are internal indicators that can be detected externally. Transgenic organisms have been produced in which the gene for the luciferase of Renilla reniformis functions stably in tobacco (Nicotiana tabaca), tomato (Lycopersicon exculentum) and potato (solanum tuberosum). Strong light emission was imaged with a low-light video camera after only a few seconds immersion of leaves, slices and seedlings (see Fig. D6.6) in 3 μmol dm-3 2-benzyl luciferin solution; at this concentration, the substrate was nontoxic and no other abnormalities were apparent[35].
Luciferase imaging enables complex gene activation effects to be modelled and observed in live animals[19]. Bioluminescent reporters for given biological processes have been used widely in cell biology; in whole animal models, including light-producing transgenic animals as models of disease, they are useful in drug discovery and development. In vivo imaging of intact organs also furthers the understanding of biological processes. The application of this technology to living animal models of infectious disease has provided insights into disease processes, therapeutic efficacy and new mechanisms by which pathogens may avoid host defences[36]. Progress of infections and efficacy of treatment are assessed by bioluminescent markers, e.g., light-emitting pathogens. Rapid, accessible high throughput screening is effective in vivo for pharmacokinetics, toxicology and target validation. Study of spatio-temporal patterns helps to characterize the site and time of action of drugs. There is also a possible clinical use in gene therapy and assessing gene vaccine delivery and efficacy.
There are several advantages in the use of in vivo imaging. In continuous monitoring each animal serves as its own control – introducing less variability than comparing groups of animals each analyzed at a different time; in addition, fewer animals are used in the experiments. Multiplex in vivo assays are also possible, using two or more reporters in the same animal. Chemiluminescence imaging is validated by the correlation of bioluminescence with culture cell counts (e.g., of E. coli). Investigations of this kind increase the number of data and can guide tissue sampling for subsequent biochemistry or histology.
There are also several drawbacks. Red and infrared light (590-800 nm) penetrates tissue well, but blue/green light (400-590nm), usually the bulk of the bioluminescence emission, is strongly attenuated. Luciferase, however, is the most widely-used reporter and has a broad emission including red light. The spatial resolution (3-5 mm) is worse than in magnetic resonance imaging or computed tomography.
Some pathological processes result spontaneously in light production. This is due to a weak spontaneous photon emission associated with oxidative phenomena, such as oxygen free radical (OFR) formation (ADD LINK), in whole organs removed from living animals. OFR have been imaged in rat livers that have been subjected to ischaemia (occlusion of blood supply) and reperfusion (restoration of blood supply), showing distribution in space and time of superoxide radicals on the liver surface and the effects on them of antioxidants, which remove OFR and can be screened by this model. The roles of aging, ethanol consumption and fat deposition on OFR formation in the liver have also been assessed. This system can be used to monitor the storage of organs for transplantation and to test agents and procedures for preserving them.