
2.1: Luminol
- Page ID
- 76102
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Luminol is the common name for 5-amino-2,3-dihydro-1,4-phthalazinedione (often called 3-aminopthalhydrazide). Oxidation of luminol produces excited 3-aminophthalate, which on relaxation emits light (λmax = 425 nm) with quantum yield of ~0.01[1]; Information on the hazards of using luminol is available at the website of the United States National Toxicology Program. The reaction is triggered by a catalytic process, usually enzymatic, provided, for example, by heme-containing proteins, especially horseradish peroxidase (HRP, EC 1.11.1.7). In the presence of hydrogen peroxide this enzyme is converted into intermediary complexes before being regenerated. It has the distinct advantage in biological work of permitting the luminol reaction at pH as low as 8.0 to 8.5. HRP can be used as a label to detect analytes of interest and luminol chemiluminescence can be used to detect substrates of oxidase enzymes that generate hydrogen peroxide. Enzymatic catalysis is discussed fully in section B1f (ADD LINK). The catalyst may be chemical rather than enzymatic (e.g., transition metal cations or complex ions, e.g., ferricyanide, at high pH). Catalysis by metal ions is discussed fully in "The role of metal ions and metallo-complexes in Luminol chemiluminescence" by HL Nekimken.
Alternatively, luminol chemiluminescence may be triggered electrochemically. Sakura[2] had proposed that luminol was oxidized at the electrode surface, after which it can react with hydrogen peroxide producing one photon per hydrogen peroxide molecule (compared with 0.5 in the HRP-catalysed reaction) giving more sensitive detection and avoiding the fragility of enzyme methods[3]. Luminol electrochemiluminescence is discussed fully in section B1d.
Very many assays have been devised determining compounds by inhibiting, enhancing or catalysing luminol chemiluminescence. Detectivity reaches the sub-femtomole level but the very versatility of the chemistry limits its selectivity. This is a serious shortcoming for samples such as body fluids or natural waters are very complex; in some cases, one analyte might enhance the luminol reaction while another inhibits it and the resulting signal is a combination of effects that is difficult to interpret. The situation is rather less difficult in process analytical chemistry, where there may be one and only one expected analyte. Coupling the chemiluminescence reaction post-column with a separation step (liquid chromatography or capillary electrophoresis) (ADD LINKS) can overcome interferences and give fmol-pmol detectivity. Labelling of sample components with luminol before separation can achieve the same end. Selectivity can also be provided by coupling the luminol reaction with enzymatic reactions or with antibody detection or with recognition by molecularly imprinted polymers[3].
Many analogues of luminol have been synthesized[1]; some of them give more intense chemiluminescence than luminol itself but only if the modifications are restricted to the benzenoid ring of the luminol molecule. Changes to the heterocyclic ring abolish chemiluminescence. Phthalic hydrazide (luminol without the amine substituent) is not chemiluminescent except in aprotic solvents.
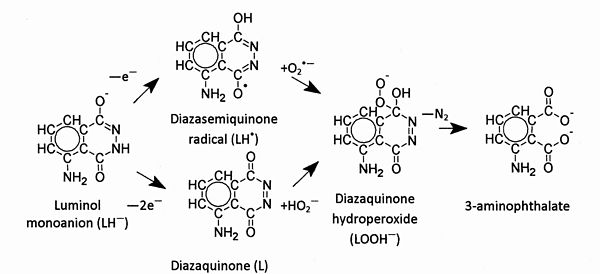
Figure B1.1 – One- and two-electron routes of primary oxidation of luminol leading to secondary oxidation and chemiluminescence.
A mechanism for the oxidative chemiluminescence of luminol has been proposed by Roswell and White[1]; some of the individual steps have been studied by Lind, Merenyi and Eriksen[4]. Figure B1.1 is a flow chart of the mechanism; the structures of the main chemical species involved in the oxidation of luminol and the abbreviations for them used in the text are shown. The model proposes two step formation of luminol diazaquinone hydroperoxide anions (LOOH–), which spontaneously decompose (via a tricyclic endoperoxide transition state) to form dinitrogen and excited 3-aminophthalate anions that luminesce. The quantum yield of luminol oxidation by this route is high giving good analytical sensitivity.
b(i) Primary oxidation of luminol
The hydroperoxide intermediate (LOOH–) is formed in aqueous solution by the primary oxidation of the luminol monoanion (LH–) to a radical (L•–) followed by the addition of superoxide (O2•–) or by primary oxidation to diazaquinone (L) followed by addition of hydrogen peroxide anions (HO2–)[5].
(a) Luminol (LH2) exists in aqueous solutions at pH 10.0 as monoanions (LH–), which undergo one-electron oxidation, e.g., by hydroxyl radicals (HO•, E0 = +2.8 V), to form rapidly (k = 9.7 x 10-9 dm3 mol-1 s-1) diazasemiquinone radicals (L•–):
1) LH– – e– → LH• (e.g., LH– + HO• → L•– + H2O)
(b) Two-electron oxidation of luminol monoanion, e.g., with hydrogen peroxide, gives diazaquinone (L),
2) LH– – 2e― → L + H+ (e.g., LH– + H2O2 → L + H2O + HO–)
Two-electron oxidation occurs at the start of the luminol-hydrogen peroxide reaction. There is no superoxide present until hydrogen peroxide, competing with luminol for the hydroxyl radical, is converted to HO2•, which rapidly deprotonates to O2•– at high pH (pKa = 4.8):
3) H2O2 + HO• → O2•– + H3O+ Hydroxyl radicals reacting with luminol convert the monoanions (LH–) to L•– or LH•, depending on the pH; this is a one-electron oxidation process. Transfer of a second electron to form diazaquinone occurs only in the absence of superoxide, which otherwise would react with L•– or LH• to form luminol diazaquinone hydroperoxide anions (LOOH–).
The primary oxidation step usually determines the rate of light emission, so luminol chemiluminescence effectively measures the power of the oxidant to bring about this reaction but other factors also affect the rate of primary oxidation. Light emission from the reaction between luminol and hydrogen peroxide can be induced by the presence of cobalt(II) at concentrations low enough to be regarded as catalytic and it has been proposed that cobalt(II)-peroxide complex ions bring about the primary oxidation of luminol[6].
b(ii) Secondary oxidation of luminol
In analytical luminol chemiluminescence, the initial oxidation of luminol is the rate-determining step. But chemiluminescence also depends on the availability of superoxide or hydroperoxide ions for secondary oxidation. So experiments have been performed using pulse radiolysis to bring about primary oxidation, allowing the rate of secondary oxidation to be studied in the pauses between the pulses. Protonated diazasemiquinone radicals (LH•) formed by one-electron primary oxidation add to superoxide radicals (O2•–) to form the diazaquinone hydroperoxide (LOOH–):
1) LH•– + O2•– → LOOH–
This reaction consumes superoxide radical anions and, in the presence of a large excess of hydrogen peroxide, the major part of LH•. LH•, however, can also recombine with itself. In the absence of superoxide, all luminol radicals are consumed by recombination, at least 80% of which is accounted for by dimerization. Diazaquinone (L), the product of two-electron primary oxidation of luminol, is converted to the peroxide by the addition of hydroperoxide anions:
2) L + HO2– → LOOH–
b(iii). Decomposition of hydroperoxide intermediate
Secondary oxidation is followed by the decomposition of the cyclic hydroperoxide intermediate to 3-aminophthalate, which emits light on relaxation to the ground state.
LOOH– → 3-aminophthalate + N2 + hν
The basic peroxide adduct (LOOH–) decomposes to form the excited state of the aminophthalate emitter, while the protonated adduct undergoes a non-chemiluminescent side reaction which forms a distinct yellow product, the so-called “dark reaction”[7]. The absorbance spectrum of LOOH– decays at the same rate as does the light emission. Emission intensity increases with pH up to a maximum at about pH = 11, reflecting increasing dissociation of H2O2 into its anion and the diminishing importance of the dark reaction . Decreased light output above pH 11 reflects diminished fluorescent quantum yield (ΦFL) of the emitter.
b(iv) Determination of chemiluminescence quantum yield
Lind and Merényi[8] have measured the light yield of several chemiluminescent reactions of luminol undergoing one-electron oxidation by hydroxyl radicals of radiolytic origin. Using the transfer of 100 eV from ionising particles to aqueous media, it becomes possible to calculate ΦCL by ΦCL = G(hν)/ ΦCLGOH. They propose as a standard for luminol chemiluminescence initiated by pulse radiolysis a system consisting of 10−3 mol dm−3 aqueous luminol at pH = 10.0 saturated with 10% O2 and 90% N2O. Having defined a standard with a well-defined light output, it then becomes possible to determine the chemiluminescence quantum yield of any other luminol reaction relative to the standard. This has been done by Merényi and Lind[9] by plotting integrated light intensity as a function of radiolytic dose (which has a linear relationship with hydroxyl radical concentration). The light yields and hence the relative quantum yields are obtained by comparing the slopes of the plots.
B1c. Oxidants used in Luminol Chemiluminescence
c(i). Hydrogen peroxide
Hydrogen peroxide is analytically the most useful oxidant of luminol, but requires the catalytic effect of an electrode, a metal ion or an enzyme. For example, it reacts readily with luminol in an aqueous medium in the presence of a cobalt(II) catalyst. This reaction is a very effective bench demonstration of chemiluminescence, using equal volumes of 0.1 mol dm−3 hydrogen peroxide and 1.0 x 10−3 mol dm−3 luminol in 0.1 mol dm−3carbonate buffer (pH between 10 and 11). Some metal ions, used to catalyse the oxidation of luminol e.g. iron(II), react with hydrogen peroxide: Fe2+ + H2O2 → Fe3+ + HO• + HO– to generate hydroxyl radicals (Fenton Reaction), which have very powerful oxidizing properties and can therefore bring about the primary oxidation of luminol. But they also react with hydrogen peroxide (equation1) and hydroperoxide ions (equation 2): 1) H2O2 + HO• → O2•– + H3O+ 2) HO2– + HO• → O2•– + H2O The consumption of hydroxyl radicals in these reactions diminishes the rate of primary oxidation, but the generation of superoxide increases the rate of secondary oxidation.
c(ii). Oxygen
The standard reduction potential (E0) of luminol radicals to monoanions (LH• + e− → LH−) has been determined to be +0.87 V[10]. Oxidation by molecular dioxygen (E0 = 1.229 V) is thermodynamically possible but in aqueous solutions, the reactivity is undetectably low at any pH (k = 10−8 dm3 mol−1 s−1) and so the reaction is not useful for primary oxidation. It is widely believed that air-saturated luminol solutions are indefinitely stable in the dark, even at pH = 14. In spite of this, the oxidation of luminol by dissolved oxygen in aqueous solutions is frequently reported. It is likely that what is referred to in these cases is oxidation by oxygen radicals, which may be formed from molecular dioxygen by suitable reductants such as metal ions. This phenomenon is discussed in chapter D10 along with other cases in which oxygen radicals act as chemiluminescence reagents (LINK).
In dimethylsulfoxide (DMSO) solutions, luminol exists as dianions and reacts with dissolved oxygen in the presence of a strong base with intense chemiluminescence[1]. The rate constant for the reaction is about 50 dm3mol−1s−1[10]; the rate constant for the corresponding reaction between oxygen and luminol dianions in aqueous alkali is 10−2 dm3 mol−1 s−1. Because the conditions of the reaction in DMSO solution are relatively simple, the phenomenon has found great favour as a demonstration [11], for a spatula measure of luminol in a bottle of alkaline DMSO will react on shaking at room temperature. However, while the oxidation of luminol in aqueous solution is very widely used analytically, there are no established analytical procedures making use of the reaction in dimethylsulfoxide, dimethylformamide or other organic solvents but the effect of a range of metal complexes on the reaction in DMSO has been investigated[12][13] The emitter (3-aminophthalate ion) tautomerizes in aprotic solvents such as DMSO to a quinonoid form that gives maximum emission at 510 nm; this tautomer is favoured by the pairing of luminol anions with metal cations (e.g., Na+ or K+). If luminol is oxidized in mixed solvents, there is less emission at 425 nm (reduced ion pairing) and more at 510 nm than in aqueous media. Also in mixed solvents there is less 425 nm emission in chemiluminescence than in fluorescence because in chemiluminescence the fraction of ion-pairs is determined by the transition-state rather than by the ground-state equilibrium as in fluorescence[1].
c(iii). Higher oxidation states of manganese
Permanganate ions are thermodynamically easily capable ((E0 = 1.70 V) of oxidizing luminol. A flow injection analysis of paracetamol in pharmaceutical preparations based on inhibition of luminol-permanganate chemiluminescence has been reported[14]. A little earlier, an imaginative biosensor for urea had been fabricated, in which ammonium carbonate generated by urease-catalysed hydrolysis was used to release luminol from an anion-exchange column to react with permanganate eluted from a second column, producing chemiluminescence[15]. A steady stream of novel applications of the luminol-permanganate system followed.
Oxidation of luminol by alkaline potassium permanganate produces manganate(VI) ions, which further react with luminol causing chemiluminescence. This phenomenon, termed by the authors ‘’’second chemiluminescence (SCL)’’’ has been applied in an assay for nickel(II) ions[16]. In a suitable flow injection manifold, dilute solutions of alkaline luminol and of aqueous potassium permanganate are mixed and allowed to react for a long enough time for the resulting chemiluminescence to drop to a stable minimum close to zero. The sample is then injected into the mixture and, if nickel ions are present, light emission recommences and rapidly rises to a well-defined peak before returning to baseline intensity. Optimum intensity of the second chemiluminescence was obtained by using a tenfold excess of luminol (over potassium permanganate) in 0.1 mol dm-3 aqueous sodium hydroxide and injecting sample at pH 5.10; linear relationship with nickel(II) concentration was established and the detection limit was 0.33 μg dm-3. Numerous divalent and trivalent metal ions and nitrate ions were found to interfere with the determination. The mechanism of the luminol-manganate(VI) chemiluminescence appears to be the same as that for other luminol oxidations, with the production of excited 3-aminophthalate ion emitting at 425 nm. But oxidations both by permanganate and manganate(VI) can lead to the formation of excited manganese(II), which would be an additional source of chemiluminescence. Unfortunately, in the work described, the chemiluminescence spectrum was observed only up to 490 nm, overlooking such possible contributions to the signal.
c(iv) Silver(III)
A fairly stable silver(III) complex anion, diperiodatoargentate(III) (DPA), [Ag (H2IO6)(OH)2]2−, can be readily synthesized [17]. A new chemiluminescence reaction between luminol and diperiodatoargentate has been observed in alkaline aqueous solution [18] [19]. The emission of light from this reaction is strongly enhanced by iron nanoparticles and the intensity is further increased by the addition of aminophylline[20]. This forms the basis for an assay in which the chemiluminescence signal has a linear relationship with aminophylline concentration in human serum over the range 1.0 x 10−8 to 2.0 x 10−6 mol dm−3. The detection limit is 9.8 x 10−9 mol dm−3. The relative standard deviation at 8.0 x 10−7 mol dm−3 is 4.8% (n = 10).
Penicillin antibiotics have also been found to enhance luminol-silver(III) complex chemiluminescence, which has formed the basis for a sensitive flow injection assay for these drugs in dosage forms and in urine samples. In optimized conditions, the detection limit for benzylpenicillin sodium is reported to be 70 ng cm−3, for amoxicillin 67 ng cm−3, for ampicillin 169 ng cm−3 and for cloxacillin sodium 64 ng cm−3[21].
The maximum wavelength of the light emitted is about 425 nm[18], which is the usual chemiluminescence from excited 3-aminophthalate, produced by luminol oxidation. This implies that the silver(III) complex is capable of bringing about both the primary and secondary oxidation of luminol as proposed by Shi ‘’et al.’’ who postulate one-electron primary oxidation of two luminol molecules by each diperiodatoargentate. Perhaps two-electron oxidation of one luminol molecule is more likely. The reduction potential of diperiodatoargentate(III) ion is +1.74 V[22], high enough for two-electron oxidation to convert water into hydrogen peroxide (ε0 = -1.763 V; Nernst equation indicates a millimolar H2O2 equilibrium concentration). This provides a possible mechanism for secondary as well as primary oxidation.
2.1: B1d. Electrochemiluminescence
The instrumentation of electrochemiluminescence (ECL) is dealt with in chapter D7. The resulting reaction pathways lend themselves to control of emission by varying the applied voltage or the electrode selected and are applicable to near- neutral (pH 8.0 to 8.5) aqueous solutions such as biological fluids, whereas luminol chemiluminescence usually occurs in strongly alkaline or non-aqueous solutions. It has been proposed that luminol is first oxidized at the electrode surface and, on subsequent reaction with hydrogen peroxide, the chemiluminescence quantum yield (see chapter A1 ADD LINK) is enhanced[23][24]. Typical of the early applications is an assay of lipid hydroperoxides using ECL at a vitreous carbon electrode[25]. With applied voltages of 0.5-1.0 V, luminol monoanion loses one electron giving diazasemiquinone, which dismutes to produce diazaquinone, which reacts quantitatively with the analyte. At applied voltages above 1.0 V, the –NH2 of diazaquinone and the analyte itself are oxidized giving respectively –NH• and superoxide, which causes an interfering signal. The detection limit in optimized conditions was 0.3 nmol at S/N = 1.5. Using a voltage of 0.5-1.0 V applied to a platinum electrode, both methyl linoleate hydroperoxide (MLHP) and luminol are oxidized; the detection limit for MLHP is 0.1 nmol at S/N = 2.5. There was no emission from the closely related methyl hydroxyoctadecadienoate (a reduction product of linoleic acid hydroperoxide). The inhibition of ECL signals from luminol oxidation can be used as a method of determination of inhibitors. A recent example is the determination of melamine in dairy products and in tableware[26]. Using low voltage scan rates in phosphate buffer at high pH, ECL is observed at 1.47 V and there is a linear (r2=0.9911) decrease of ECL proportional to the logarithm of the melamine concentration over the range 1 to 100 ng cm−3. The limit of detection is 0.1 ng cm−3 with high recovery. The signal arises from the reaction with luminol of reactive oxygen species (from the electrooxidation of hydroxyl ions) that are eliminated by melamine. Modification of electrodes is now a well-established way of controlling ECL and in recent years the use of nanomaterials for this purpose has grown in importance. An example involving luminol is the modification of a gold electrode by applying a composite of multi-wall carbon nanotubes and the perfluorosulfonate polymer Nafion[27]. In the course of cyclic voltammetry in carbonate buffer, three ECL peaks were obtained, up to 20 times as intense as with the unmodified electrode; in each case the emitter was identified as 3-aminophthalate anion, indicating that the improvement was due to electrode efficiency rather than to any change in the chemistry of the system.
ECL immunosensors have been fabricated that have been successfully applied to the determination of human immunoglobulin G (hIgG) in serum. The primary antibody, biotin-conjugated goat anti-hIgG, is first immobilized on an electrode modified with streptavidin-coated gold nanoparticles (AuNPs). The sensors are sandwich-type immunocomplexes formed by the conjugation of hIgG to a second antibody labelled with luminol-functionalized AuNPs. ECL is generated by a double potential step in carbonate buffer containing 1.0 mmol dm−3 hydrogen peroxide. Many luminol molecules are attached to the surface of each AuNP and act as multiple sources of light emission from each antibody molecule. The amplification of ECL in this way, linked to the analyte by the biotin-streptavidin system, leads to greatly enhanced signals. The limit of detection is 1 pg cm−3 (at S/N = 3), which surpasses the performance of all previous hIgG assays[28].
The surface of a glassy carbon electrode was modified by producing on it L-cysteine reduced graphene oxide composites onto which AuNPs were self-assembled. Cholesterol oxidase (ChOx) was subsequently adsorbed on the AuNP surface to form a cholesterol biosensor with satisfactory reproducibility, stability and selectivity. The AuNPs increase the surface area of the electrode, hence permitting a higher ChOx loading, and provide a nanostructure more favourable to ECL, improving analytical performance. The linear response to cholesterol of the sensor extends over the range 3.3 x 10−6 to 1.0 x 10−3 mol dm−3 and the limit of detection is 1.1 x 10−6 (at S/N = 3)[29].
A poly(luminol-3,3',5,5'-tetramethylbenzidine) copolymer manufactured by electropolymerization on screen-printed gold electrodes greatly improves the ECL of hydrogen peroxide. A cholesterol biosensor suitable for the analysis of serum samples was fabricated by immobilization of cholesterol oxidase onto the polymer. Under optimized conditions, the biosensor has a linear dynamic range of 2.4 x 10−5 to 1.0 x 10−3 mol dm−3 with a limit of detection of 7.3 x 10−6 mol dm−3. Precision (measured as relative standard deviation) was 10.3% at 5.0 x 10−4mol dm−3 and the method has the additional advantages of low cost and high speed[30].