
4: ab initio Calculations - Electron-Electron Repulsion (Dry Lab)
- Page ID
- 65238
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Name: ______________________________
Section: _____________________________
Student ID#:__________________________
Template:HideTOCWe will use the cool web base ab initio site by Perri at Sonoma State U. Download the following paper and review the concept (http://pubs.acs.org/doi/pdf/10.1021/ed5004228). Follow the directions on this tutorial (you will not need to install Avogadro for this HW). You can complete the online tutorial if you are confused.
- click here: https://chemcompute.org
- sign in: Username: "Chem110A" and password: "Halloween"
- click on GAMESS at the top
- click on red/blue molecular orbital (under GAMESS)
- click on "submit" on top right
We are going to calculate the binding energy of the first two rows of atoms with the ChemCompute software. Before beginning, we need to discuss an advanced aspect of electronic wavefuctions: Spin. You know from your general chemistry class that electrons have a "spin" that is related to the \(m_s\) quantum number that can be either "up" with \(m_s = +1/2\) or "down" with \(m_s = -1/2\). Review your electron configuration rules (especially Hund's rules) if you are not familiar with this.
The multiplicity of an energy state is defined as \(2S+1\), where \(S\) is the total spin angular momentum and is determined by adding the \(m_s\) quantum numbers of all electrons in the system (atom or molecule):
\[S =\sum_i^n m_s(i)\]
where \(n\) is the number of electrons in the atom and \(m_s(i)\) is the \(m_s\) quantum number for the ith electron.
The spin multiplicity of the system will be discussed in detail later on in course. States with multiplicity 1, 2, 3, 4, 5 are respectively called singlets, doublets, triplets, quartets and quintets. The triplet state indicates that the multiplicity \(2S+1 = 3\), so that the total spin \(S = 1\). This spin is due to two unpaired electrons, as a result of Hund's rule which favors the single filling of degenerate orbitals.
For example, the electronic configuration of the ground state of the nitrogen atom is \(1s^22s^22p^3\) and has one electron in each of the 2p orbitals (each with the same spin so same \(m_s\) value, e.g., +1/2. So for this system
\[S = \underbrace{+ ½ -½}_{1s^2} + \underbrace{+½ - ½}_{2s^2} + \underbrace{+½ + ½ +½}_{2p^3} \,\,= 3/2\]
and the multiplicity is \(2 \times 3/2 +1 = 4\); the ground state nitrogen atom is a quartet state.
Closed Shell vs. Open Shell Systems
A system is a closed shell system if each electron is paired with another in the same orbital. Closed shell systems are singlets. The alternative is an open shell system.
You will want to prepare the table below with the appropriate details before you do the quantum calculations.
It is VERY useful to review these two pages to interpret your results for credit
The Atomic Orbital Concept and Slater's Rules
Building the first Two rows of the Periodic Table
Run the single-point calculations on the first ten atoms of the periodic table (Hydrogen to Neon) and fill in the below table. You only need to extract the total energy (binding energy) of each system for the table, but you need to address the spin of the system explicitly (those are the rules of quantum calculations).
- If the spin multiplicity of the atom is not 1, then use the UHR (Unrestricted Hartree-Fock) method,
- If the spin multiplicity of the atom is 1, you can use either the RHF (Restricted Hartree-Fock) or UHR method (should give the same results).
Use the 6-311G** basis set for both. If you select the RHF and have a non-unity spin multiplicity, the program will give you an error.
Pull in the 03166 input file (job# 3166) to get the helium atom. The hydrogen atom is already done for you
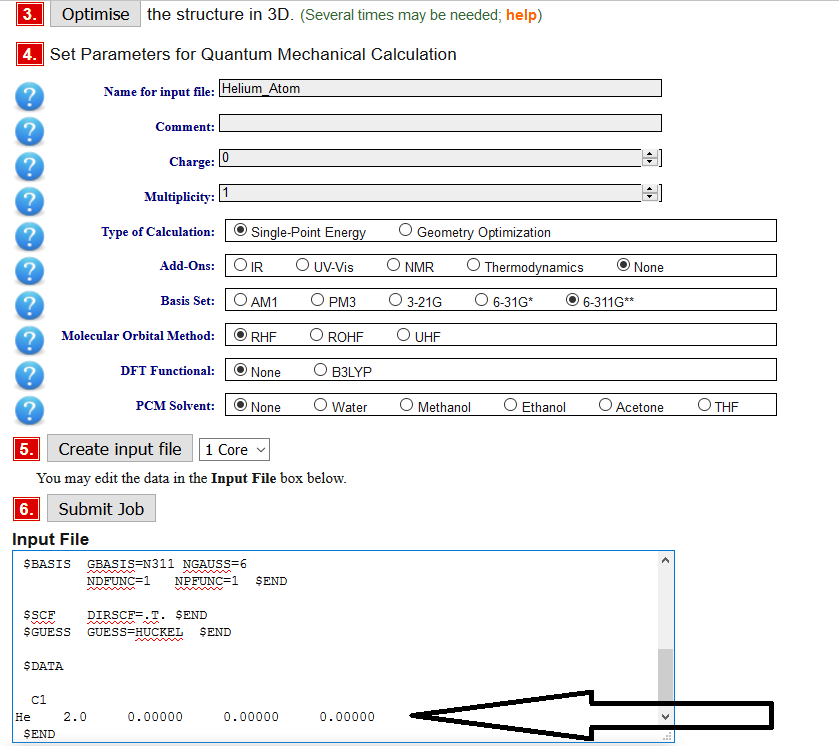
When you recalculate a new atom you will have to change the multiplicity for the molecule (from the table you are making below) and then you "create" the input file and swap to the next atom in the input (see arrow above) by changing these two aspects:
- the name of the atom to the standard chemical abbreviation and
- the number after that is the atomic number \(Z\).
| Atom | Nuclear Charge (\(Z\)) | Number of Electrons \(n\) | Electronic Configuration | Total electron spin \(S = \sum m_s\) | Spin Multiplicity \(2S+1\) | Binding Energy \(E_Z\) | \(n Z^2 E_{1} \) |
|---|---|---|---|---|---|---|---|
| Hydrogen | 1 | 1 | \(1s^1\) | 1/2 | 2 | -313 | -313 |
| Helium | 2 | ||||||
| Lithium | 3 | ||||||
| Beryllium | 4 | ||||||
| Boron | 5 | ||||||
| Carbon | 6 | ||||||
| Nitrogen | 7 | ||||||
| Oxygen | 8 | ||||||
| Fluorine | 9 | ||||||
| Neon | 10 |
Submit for Credit the Following Items or Answers
Submit the above table filled out for the total binding electronic energy of each atom (do not copy from others as if we will compare tables)
- Plot up this binding energy \(E_Z\) vs. \(n\) on your favorite software (e.g., Excel or matlab or Plot2 for mac users)
- Plot up the \(nZ^2E_1\) vs. \(n\) on same plot
- From these data, answer these questions:
- What is the definition of binding energy (i.e., total electronic energy) of these atoms?
- Why are your binding energies negative?
- What is the meaning of \(nZ^2E_{1}\) expression?
- What is the origin of the \(n\) in this expression?
- What is the origin of the \(Z^2\) in this expression?
- What is the origin of the \(E_{1}\) in this expression?
- What is the origin of difference between \(nZ^2E_1\)and \(E_n\)?
We can make an effective nuclear charge \(Z_{eff}\) that can be added to this expression such that this equation holds\[E_z = n Z_{eff}^2 E_{Z=1} \]Calculate the \(Z_{eff}\) for each of the ten atoms and plot up \(Z_{eff}\) vs. \(Z\). Explain the origin of this difference (use may want to reference Slater's Rules). Note that this is an average \(E_{eff}\) over all electron, but it normally used for a specific electron in the system.