
Chapter 14 Solutions
- Page ID
- 1112

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In-chapter exercises
E14.1:
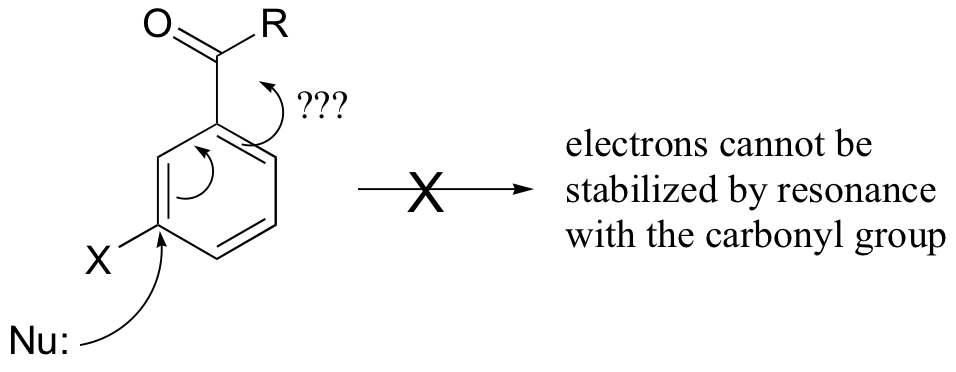
E14.2:
a)
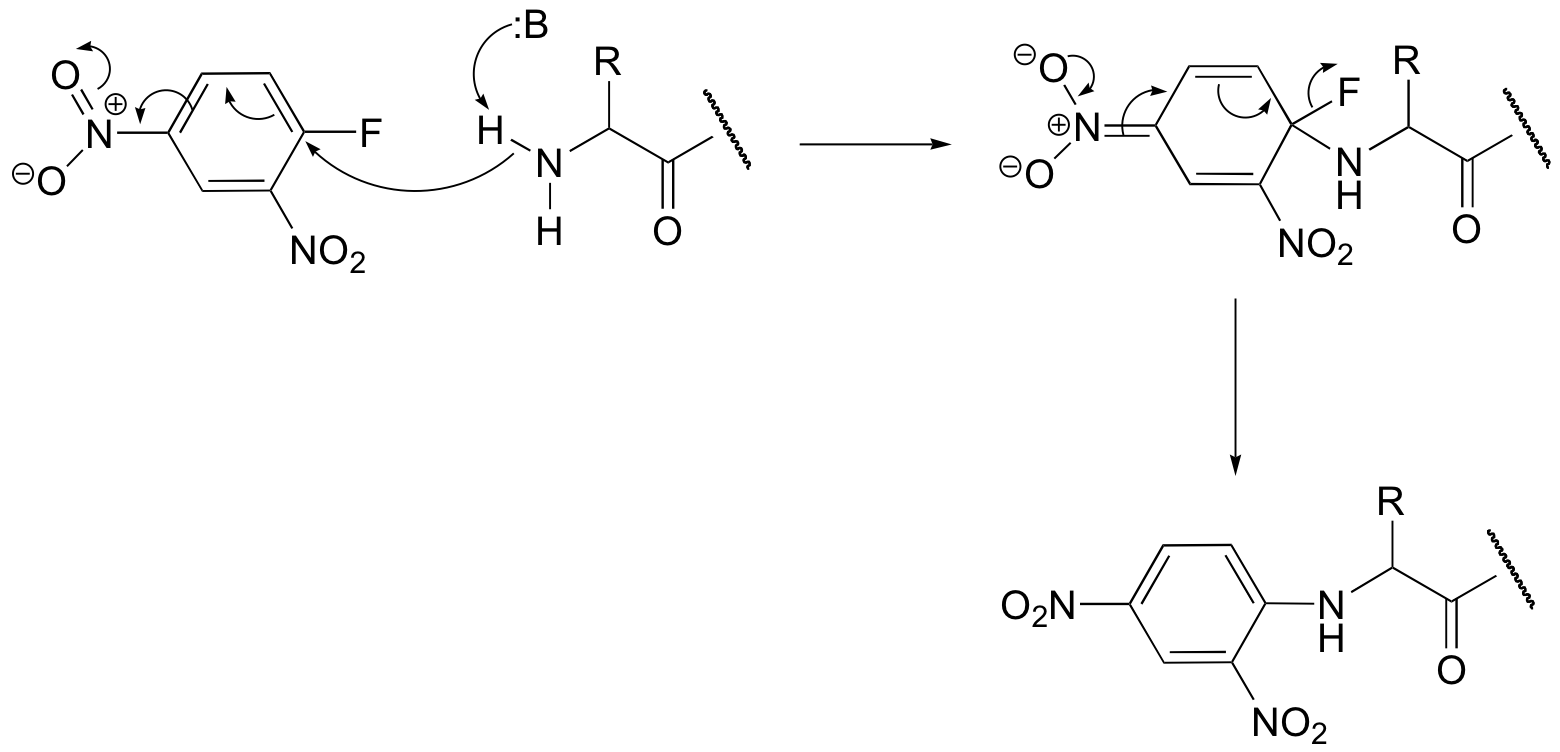
b) The second nitro group further stabilizes the negatively-charged intermediate (the negative charged can be delocalized to either one of the nitro groups).
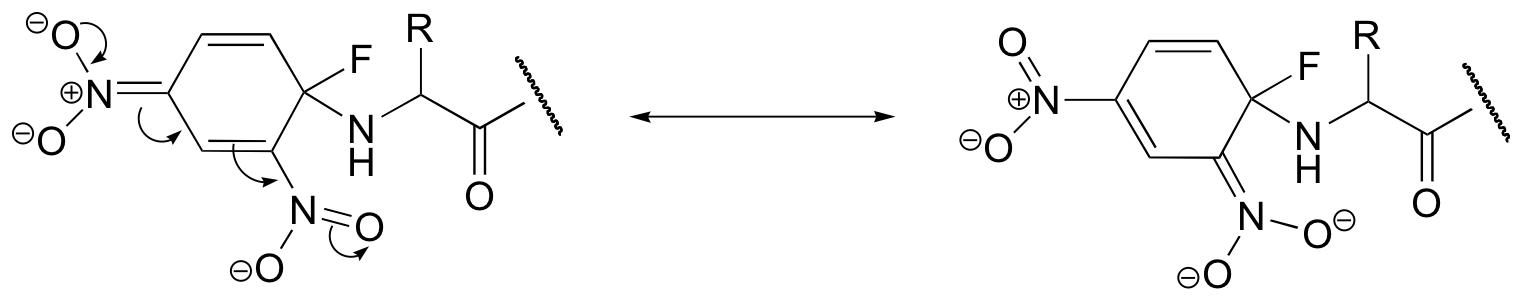
c) The step in which the halogen leaves (the second step) is NOT the rate-determining step in this reaction – the rds is the initial attack by the nucleophile (which temporarily disrupts the aromaticity of the ring). Thus the fact that fluorine is a relatively poor leaving group is inconsequential. A fluorine substituent actually works better than other halogens because it is more electronegative, making the attached carbon more electron-poor and thus a better nucleophile, increasing the rate of the rds.
E14.3:
E14.4:
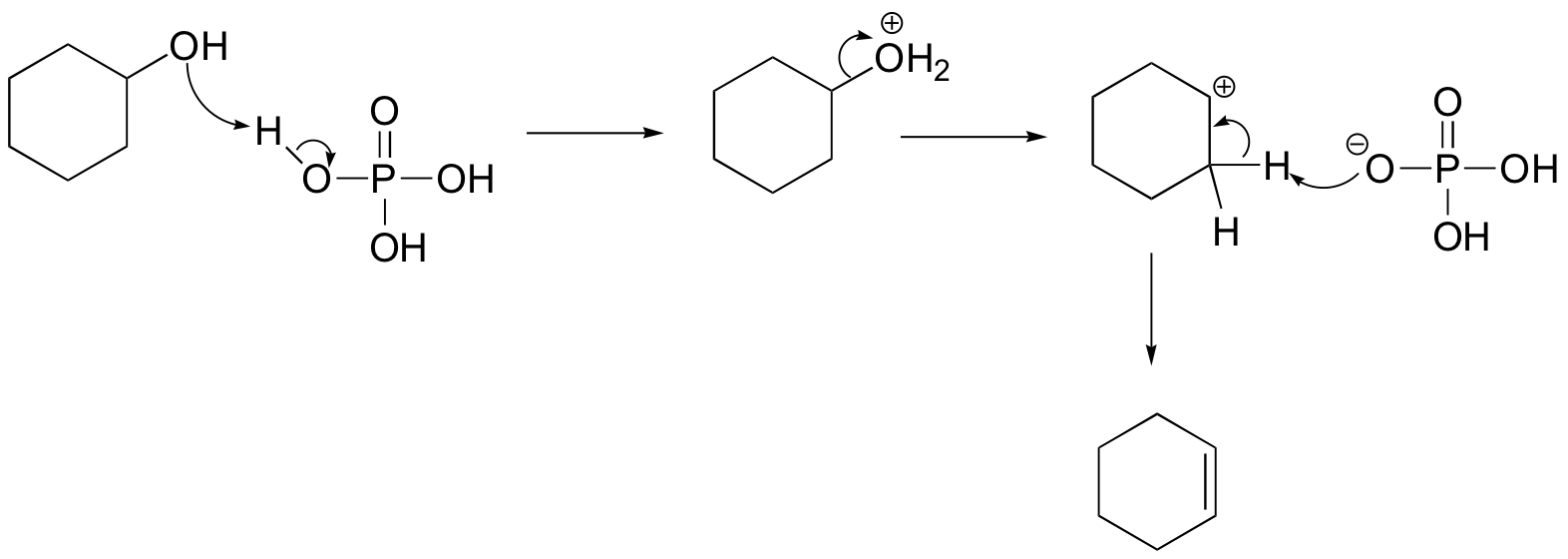
The cyclohexyl phosphate could form if the phosphate attacked the carbocation intermediate as a nucleophile rather than as a base:
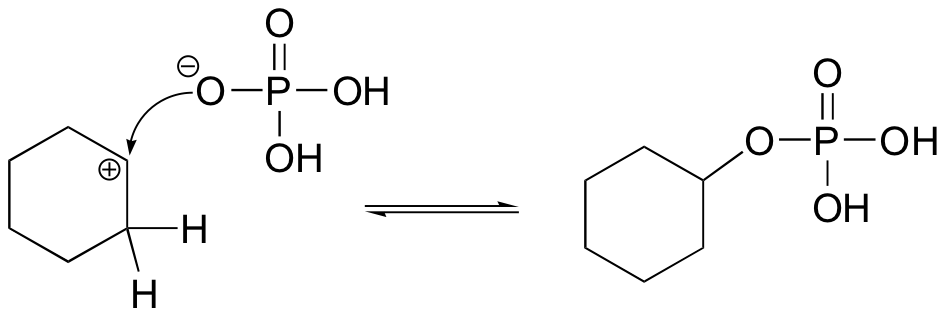
E14.5:
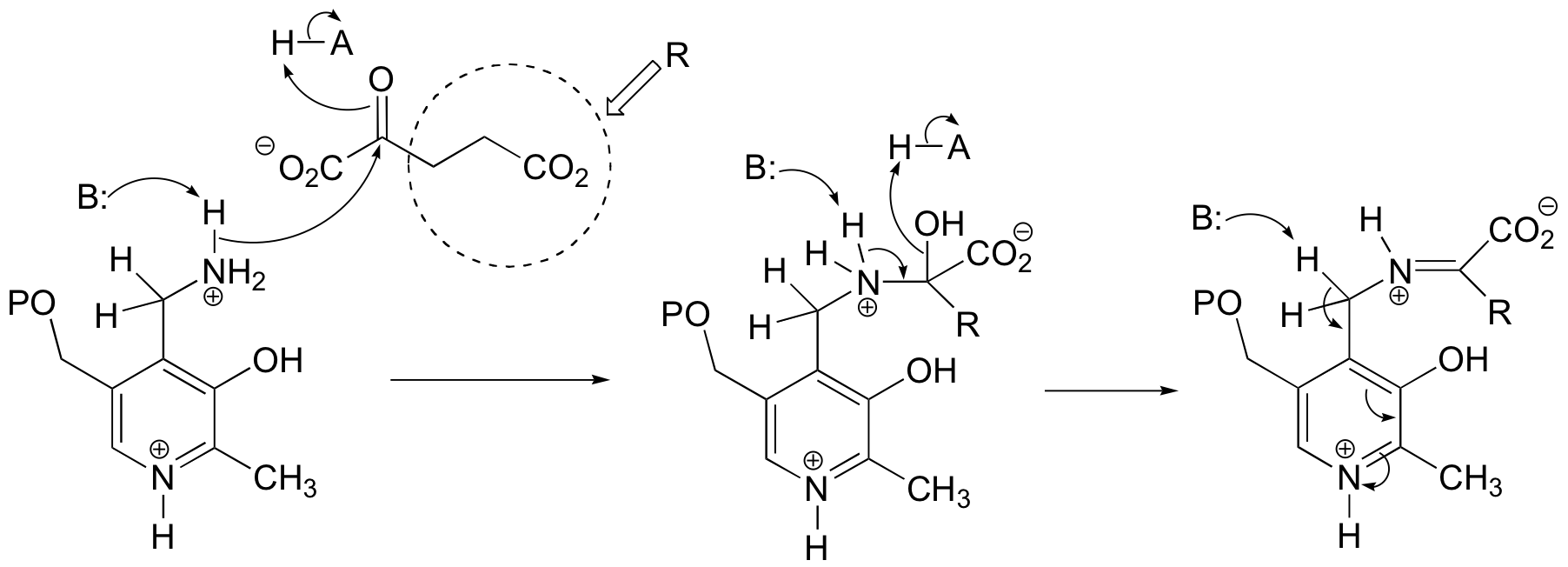
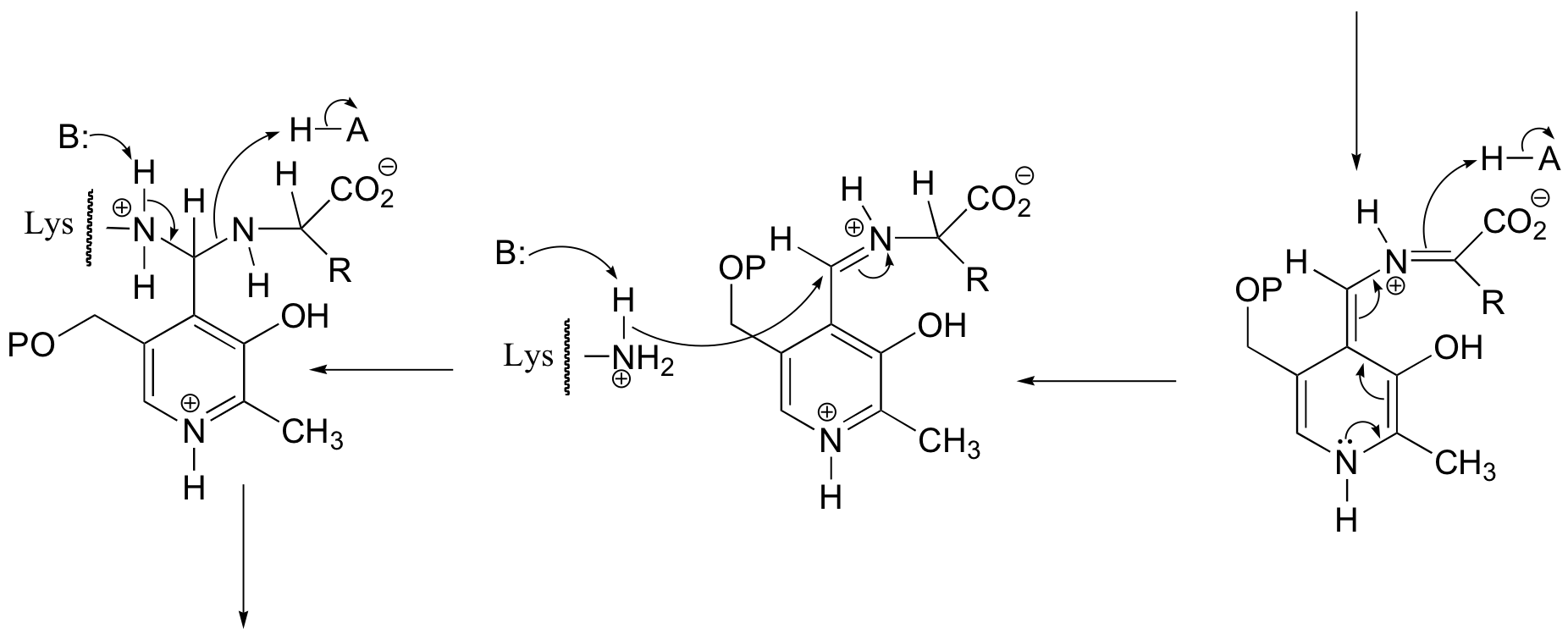
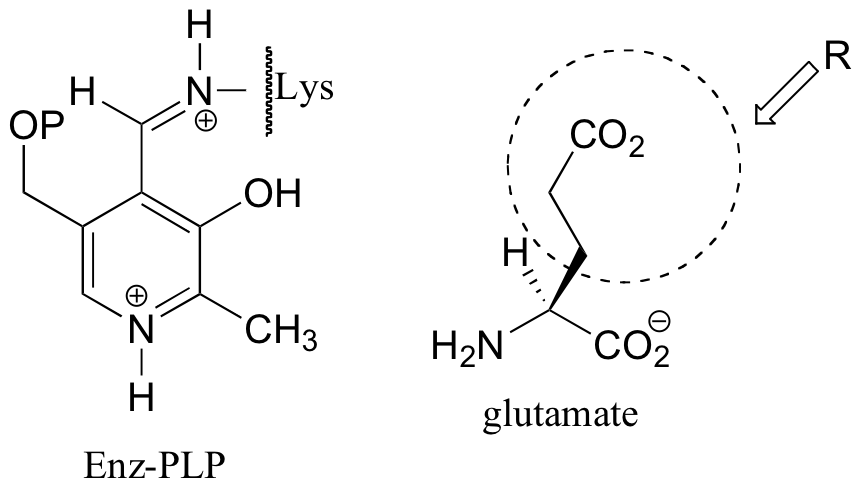
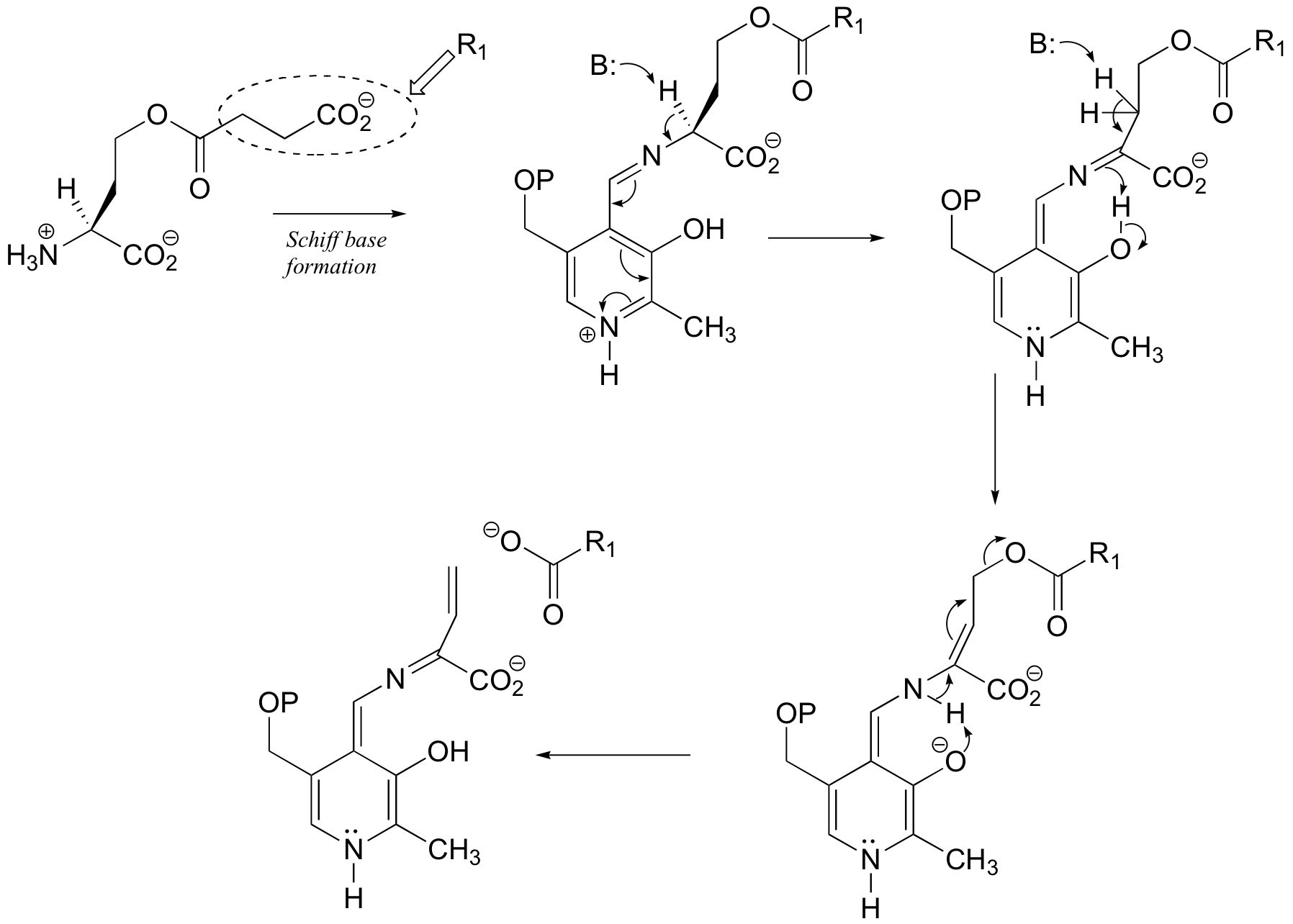
E14.6:
The elimination phase of the gamma-substitution is:
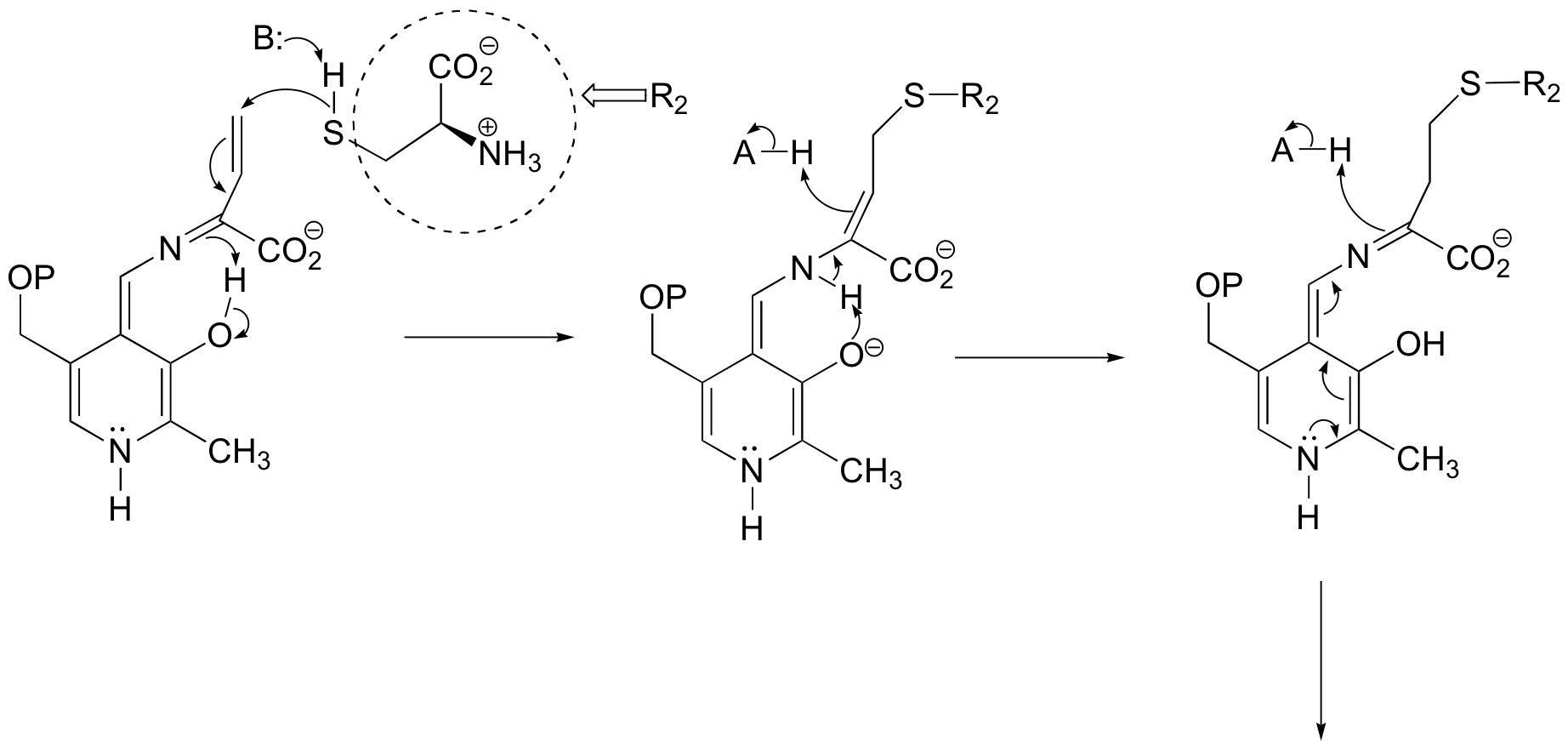
The addition phase completes the substitution:
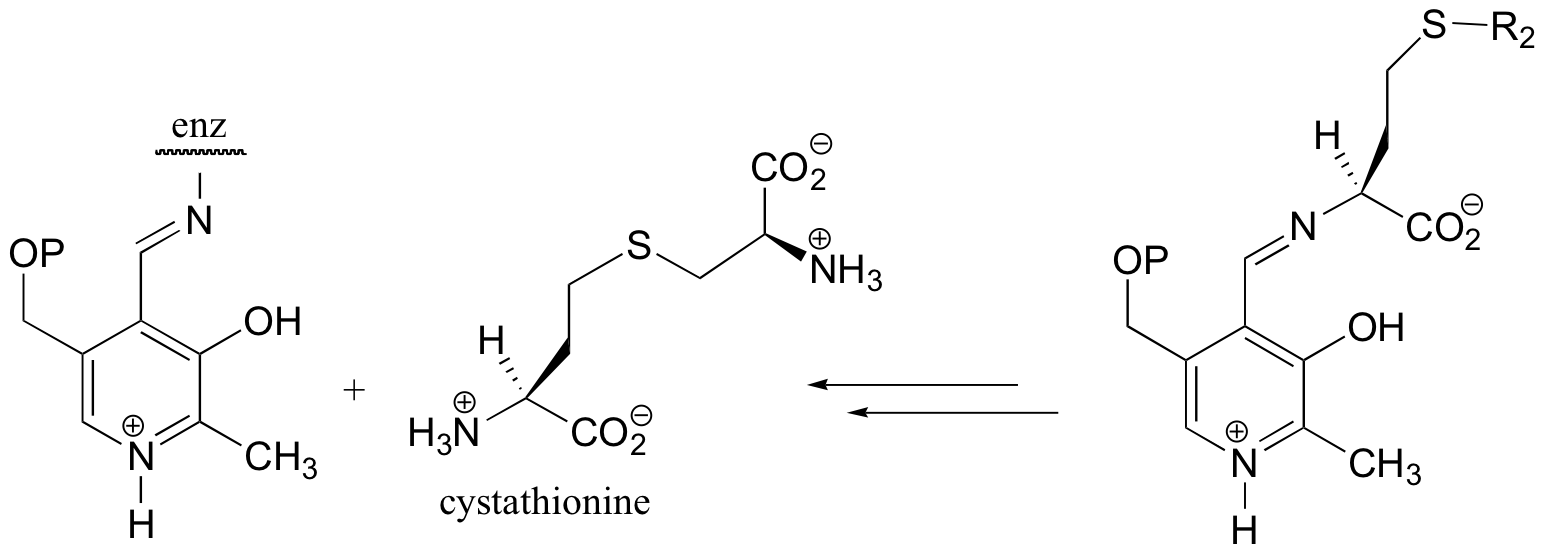
E14.7:

E14.8:
The mechanism for the hydrolysis reaction can be depicted:
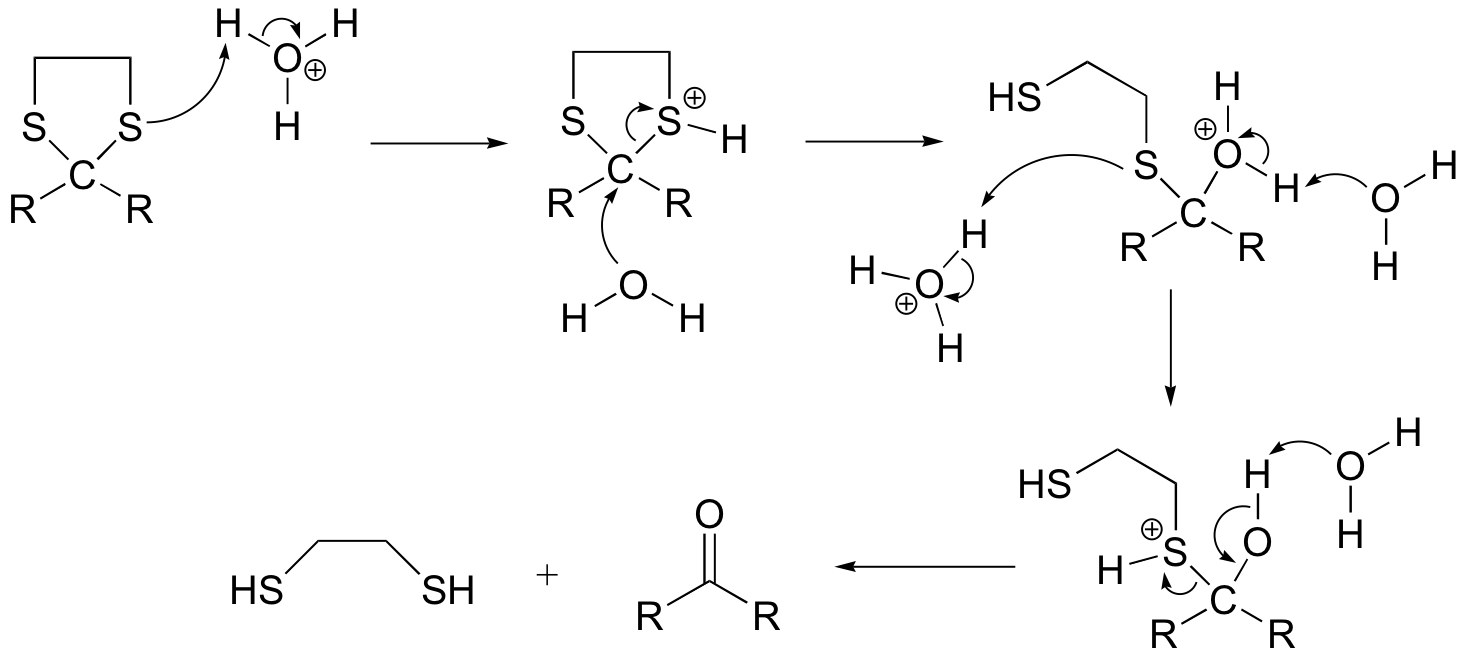
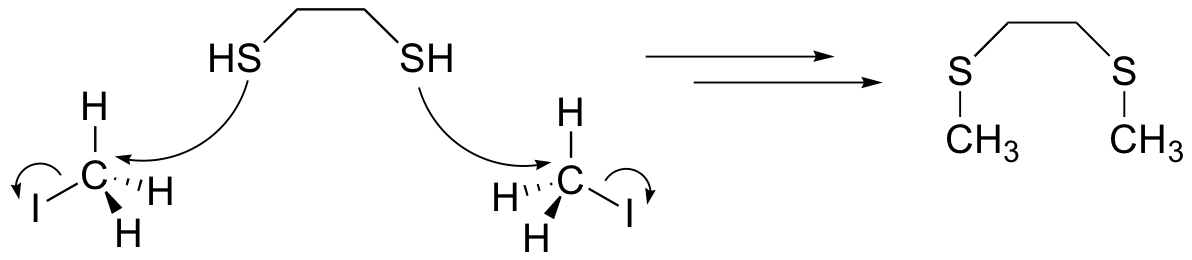
The methyl iodide helps to prevent the reverse reaction (thioacetal formation) by serving as an electrophilic 'trap' for the thiol groups:
End-of-chapter problems
P14.1: (See Biochemistry 1997, 36, 12526)


P14.2: This reaction is an elimination followed by enamine to imine tautomerization followed by hydrolysis of the imine.
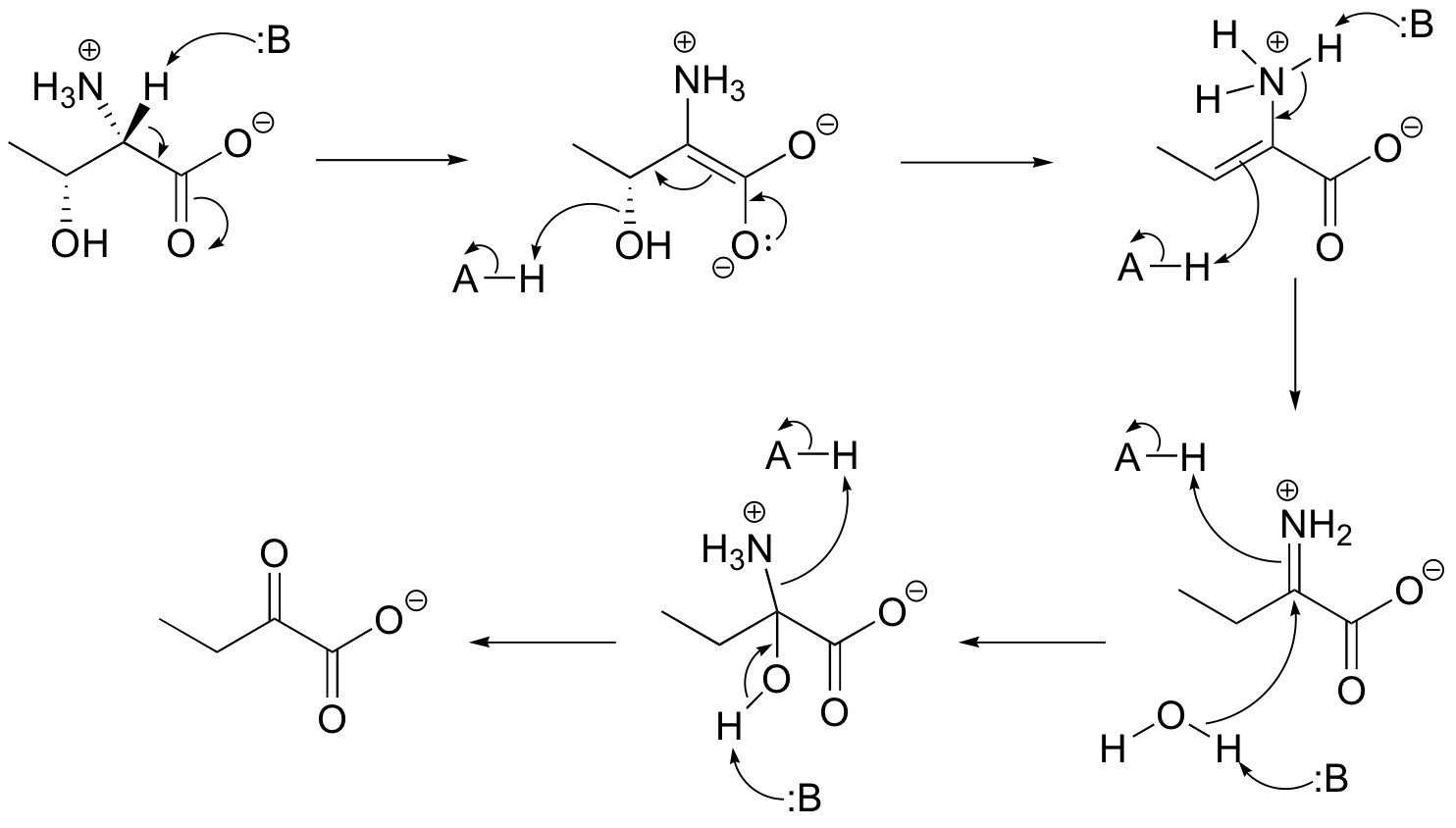
P14.3:
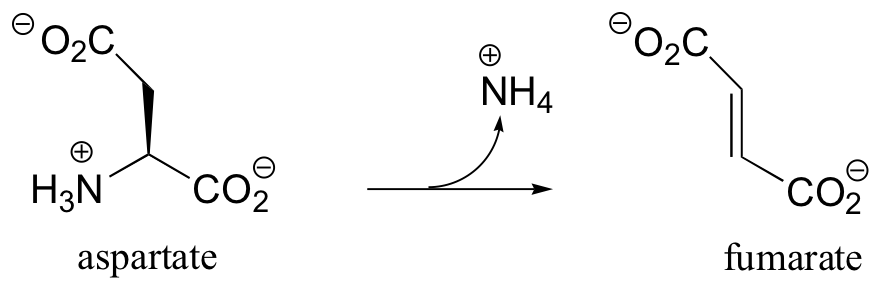
fig 5
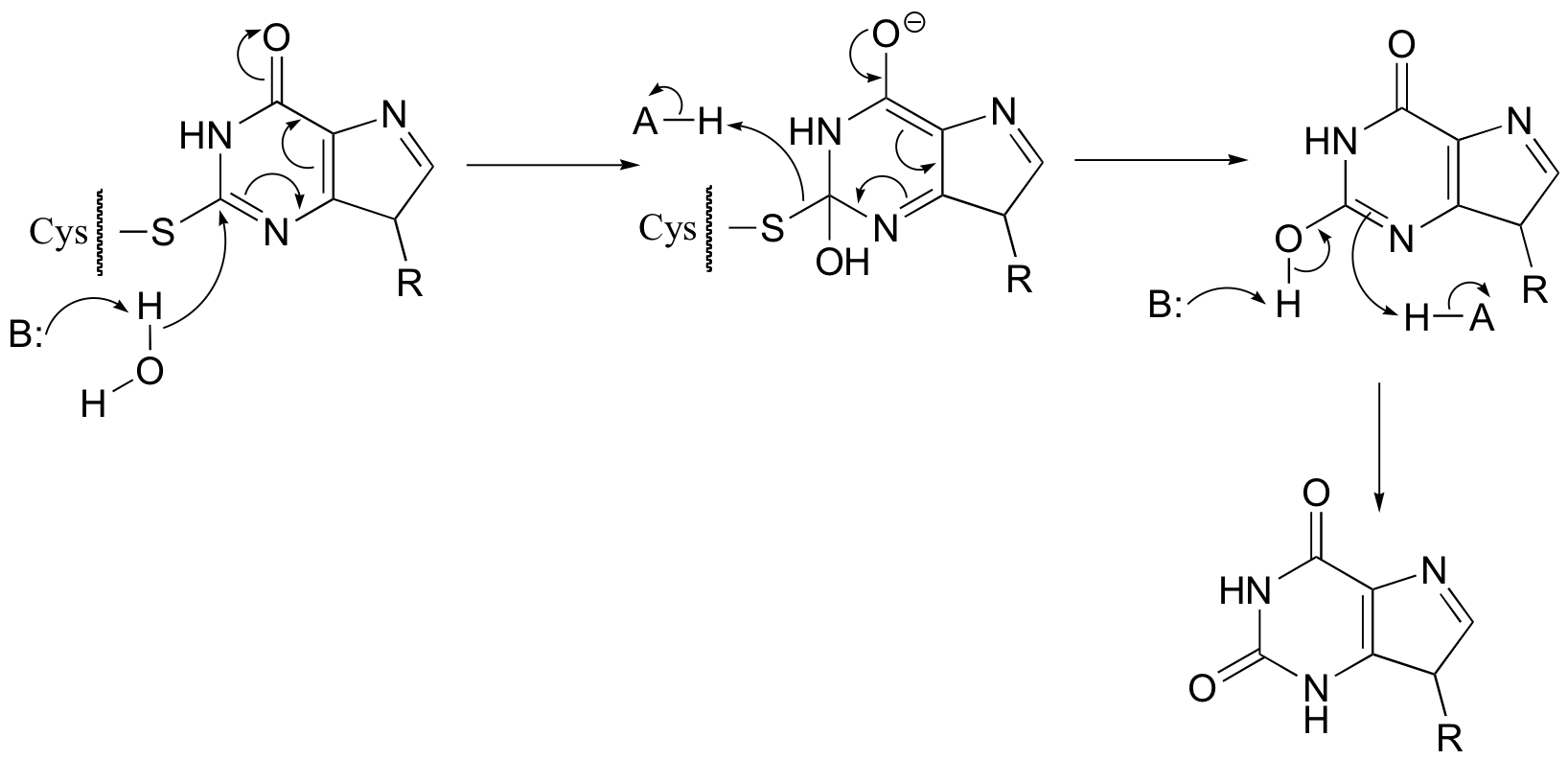
P14.4: A likely mechanism for cysteine 'tagging' is by a Michael addition.Michael addition)
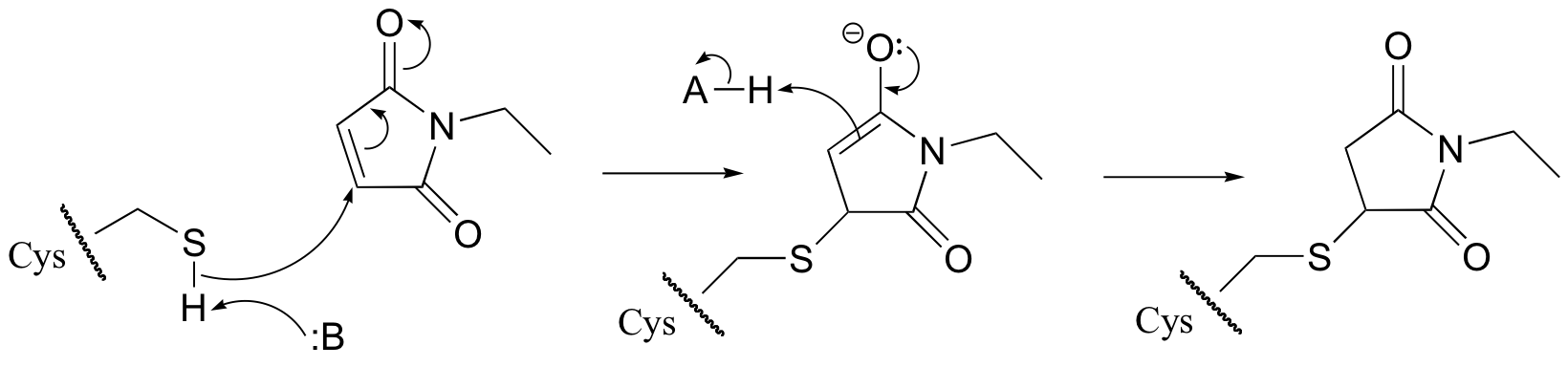
P14.5:
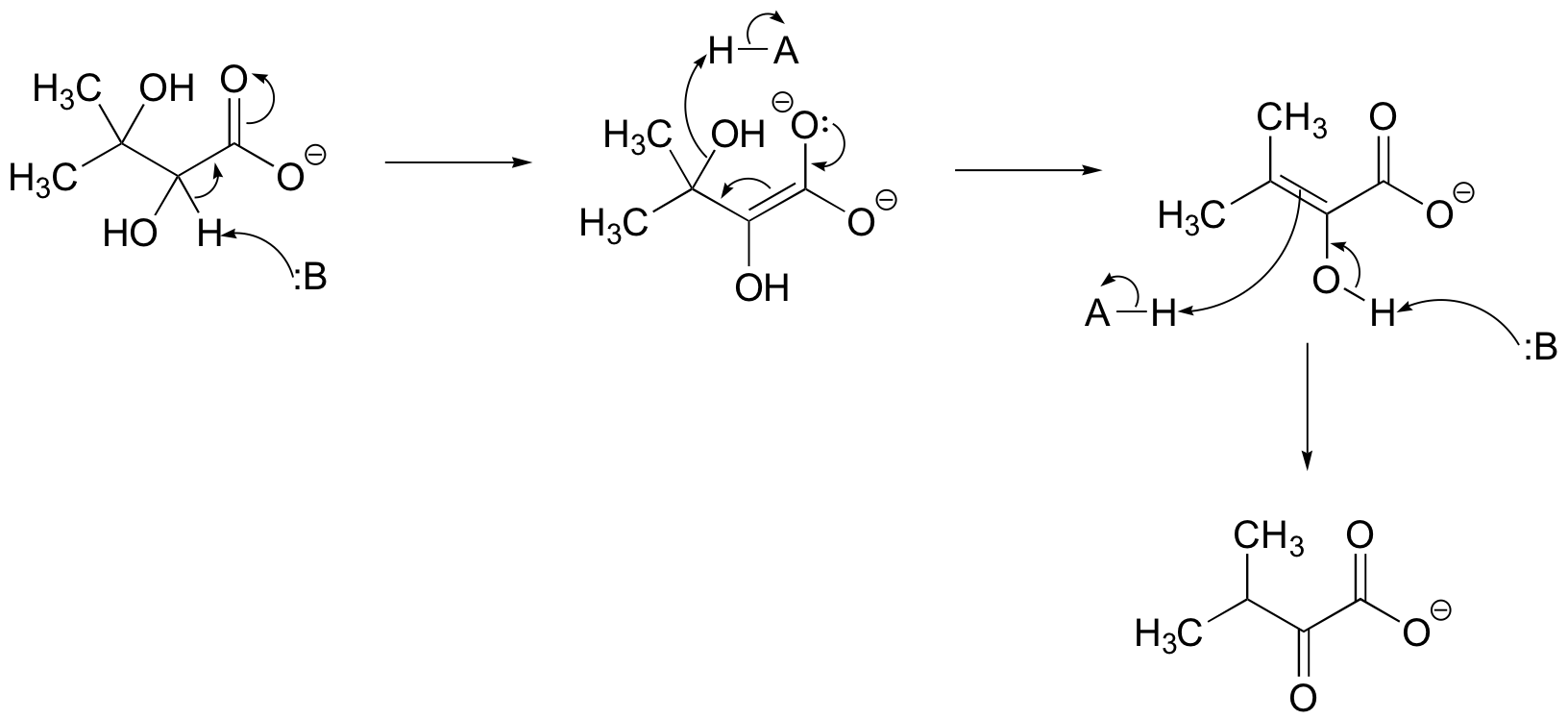
a) This is a dehydration followed by tautomerization.
b) This reaction requires thiamine diphosphate.
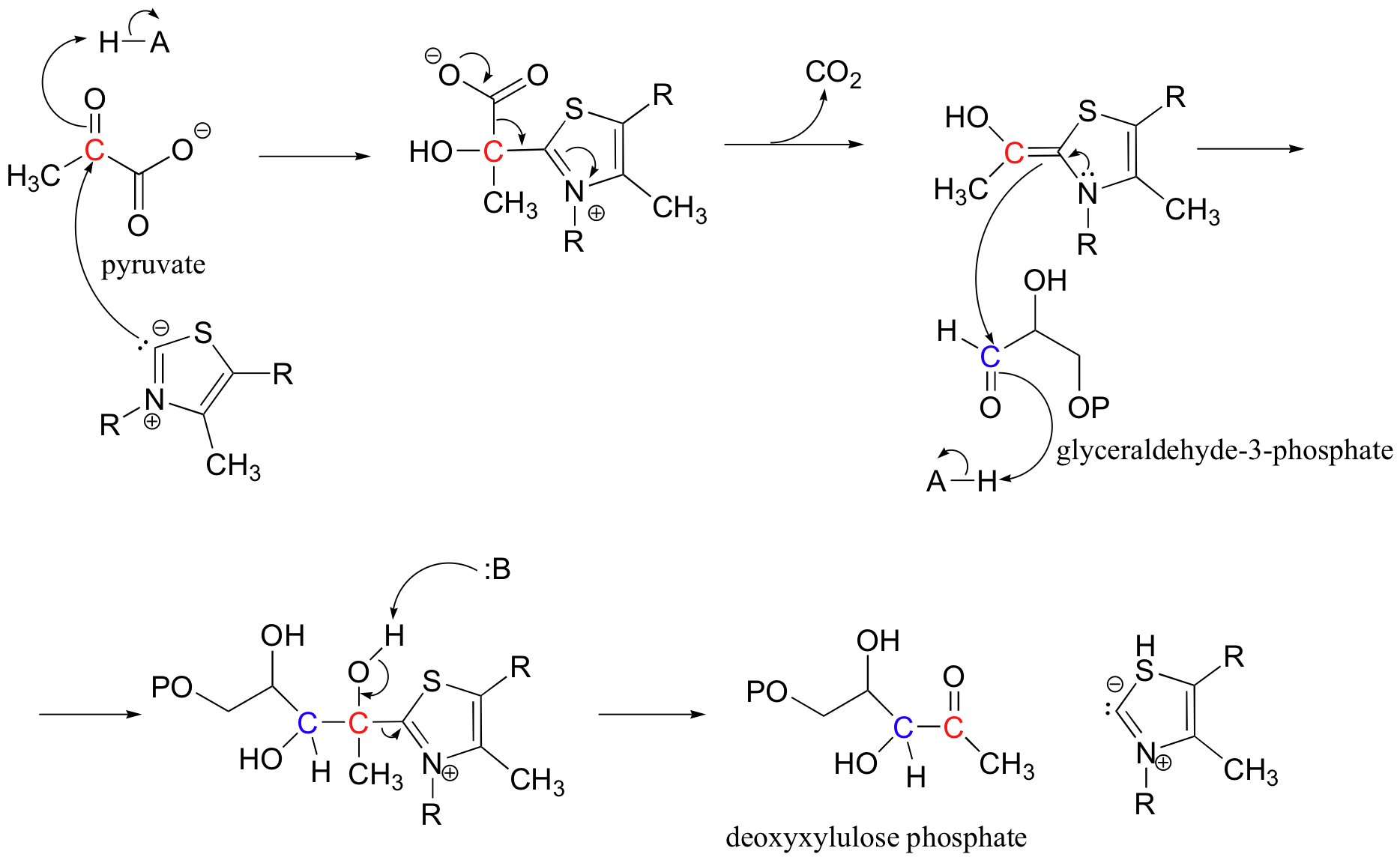
P14.6:
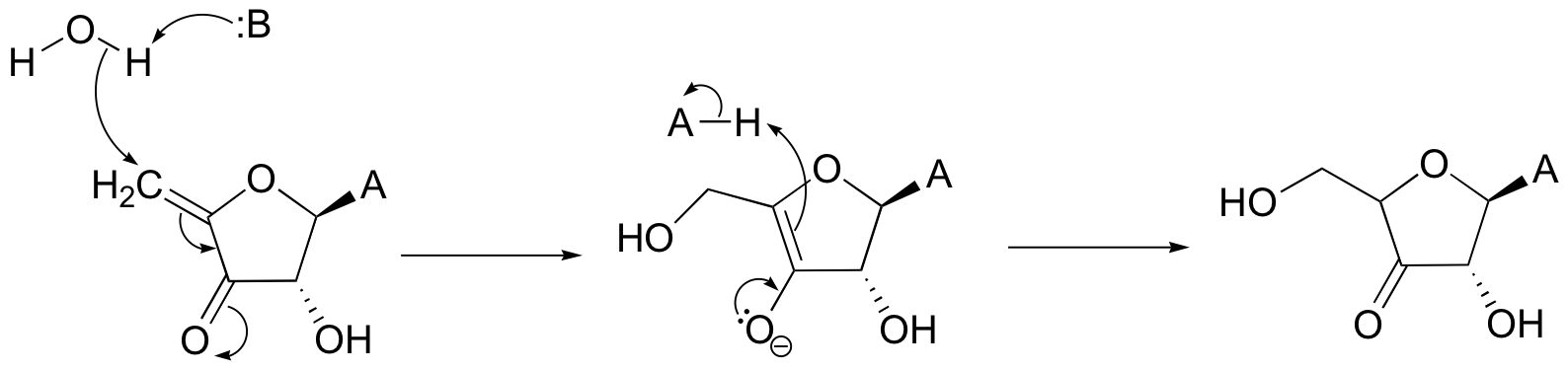
P14.7:
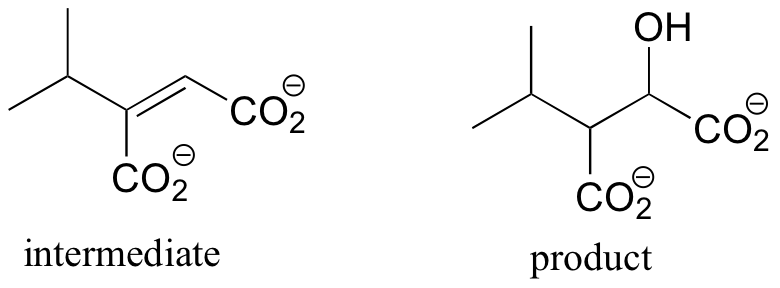
P14.8: Notice that in both reactions the more common E1cb mechanism is not possible, as there is no way to stabilize a carbanion intermediate. However, carbocation intermediates can be stabilized by resonance in both a) and b).
a) Here, the intermediate carbocation is conjugated (allylic) to the two double bonds in the ring.
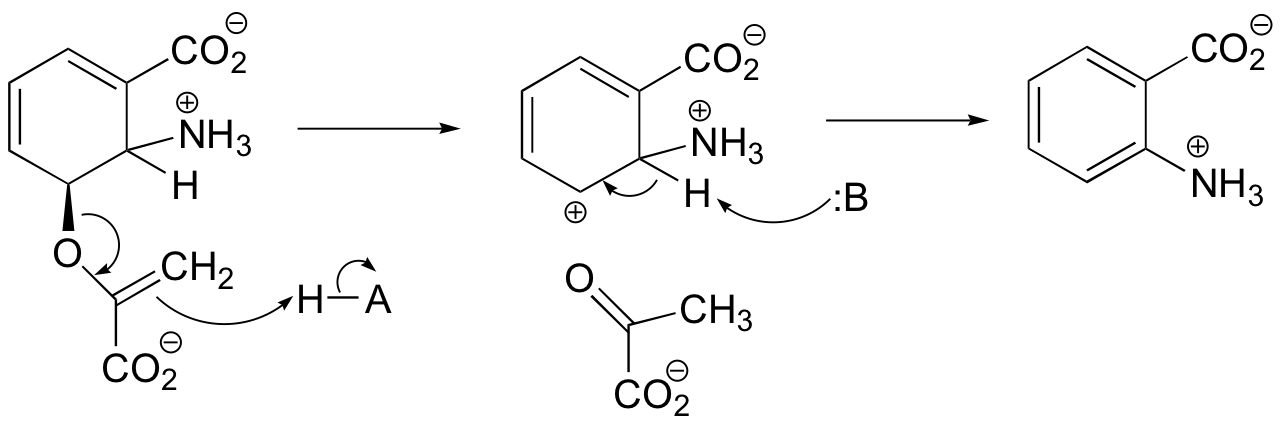
b) Here, the intermediate carbocation is stabilized by electron donation from the adjacent nitrogen.
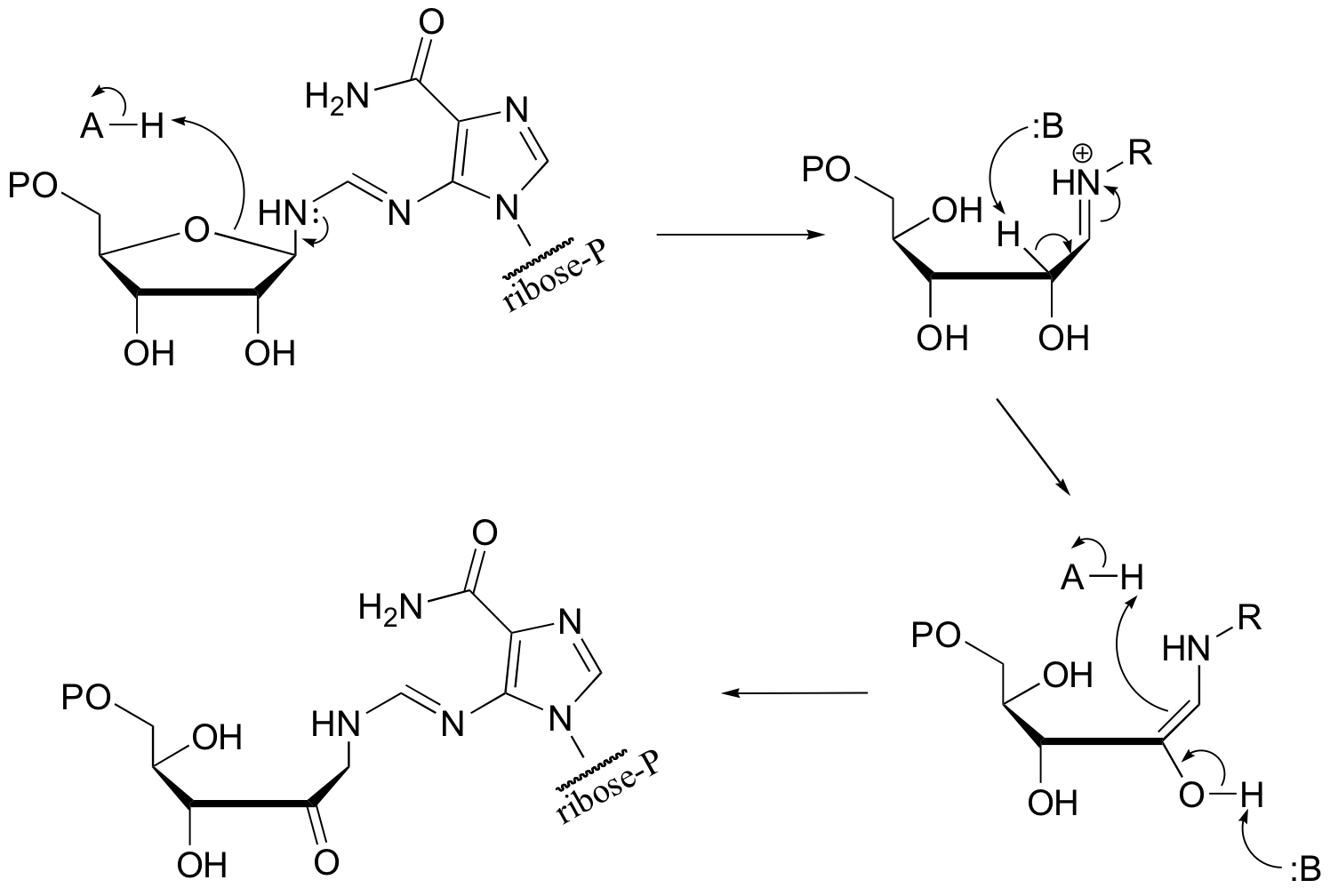
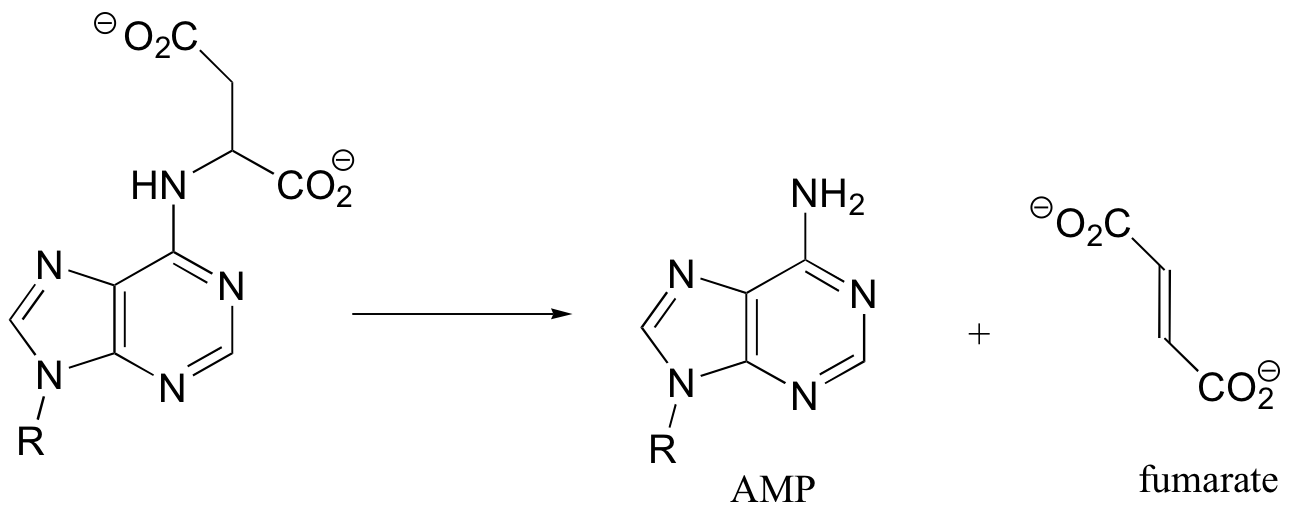
P14.9: This is an E1cb elimination - AMP is the leaving group.
P14.10:
a) This is a retro-aldol reaction.
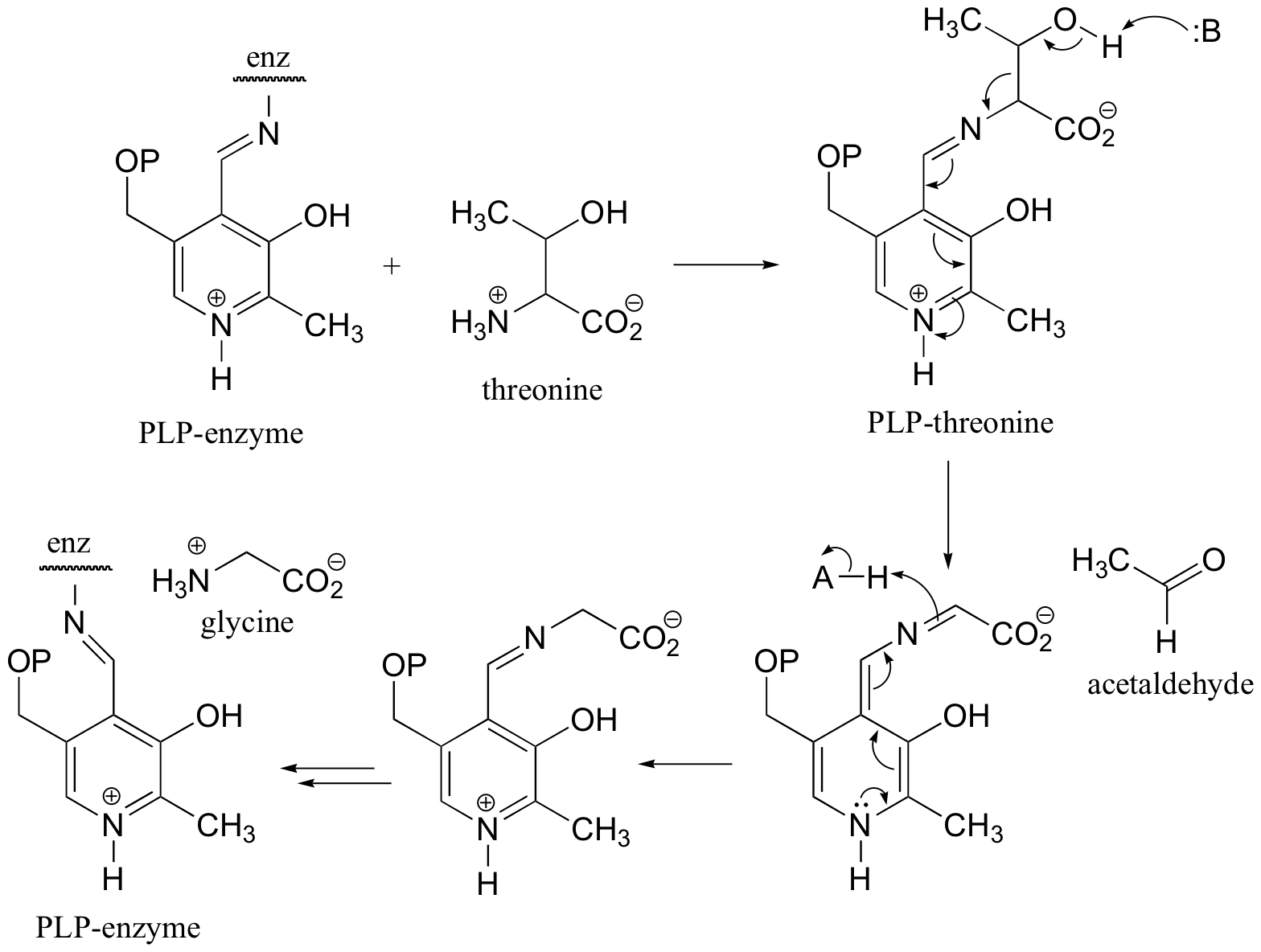
b) This is something we haven't seen before - a PLP-dependent retro-Claisen. Notice that a carbon-carbon bond is broken.
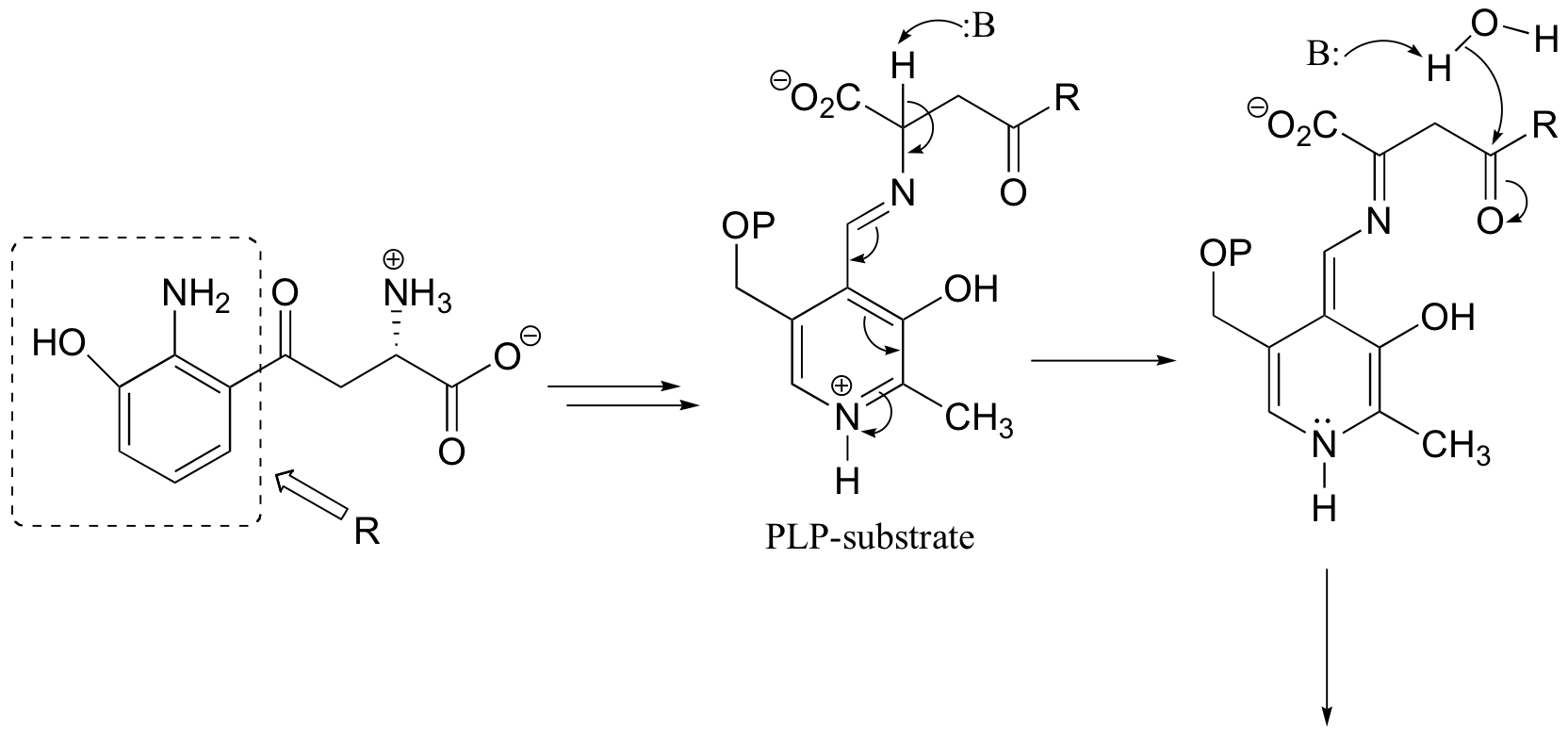
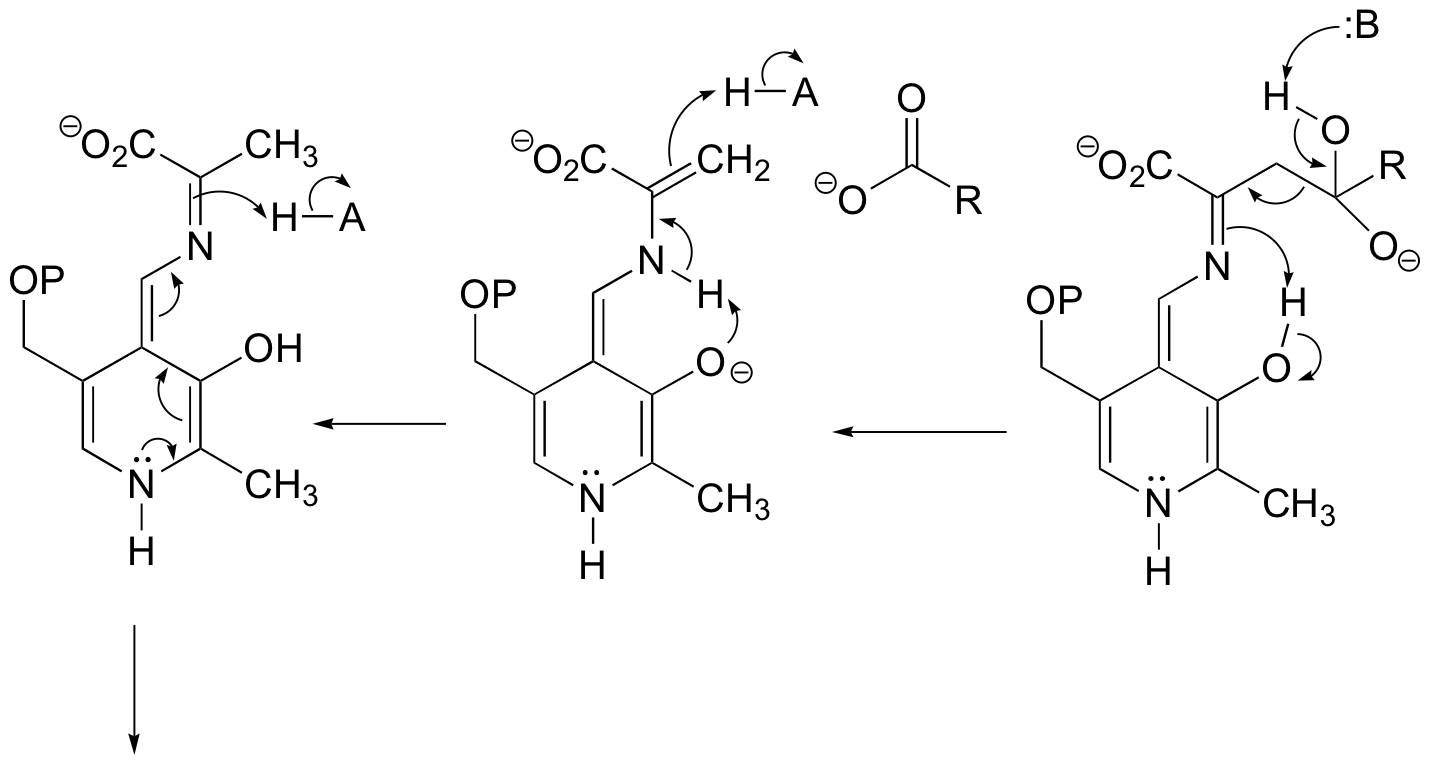
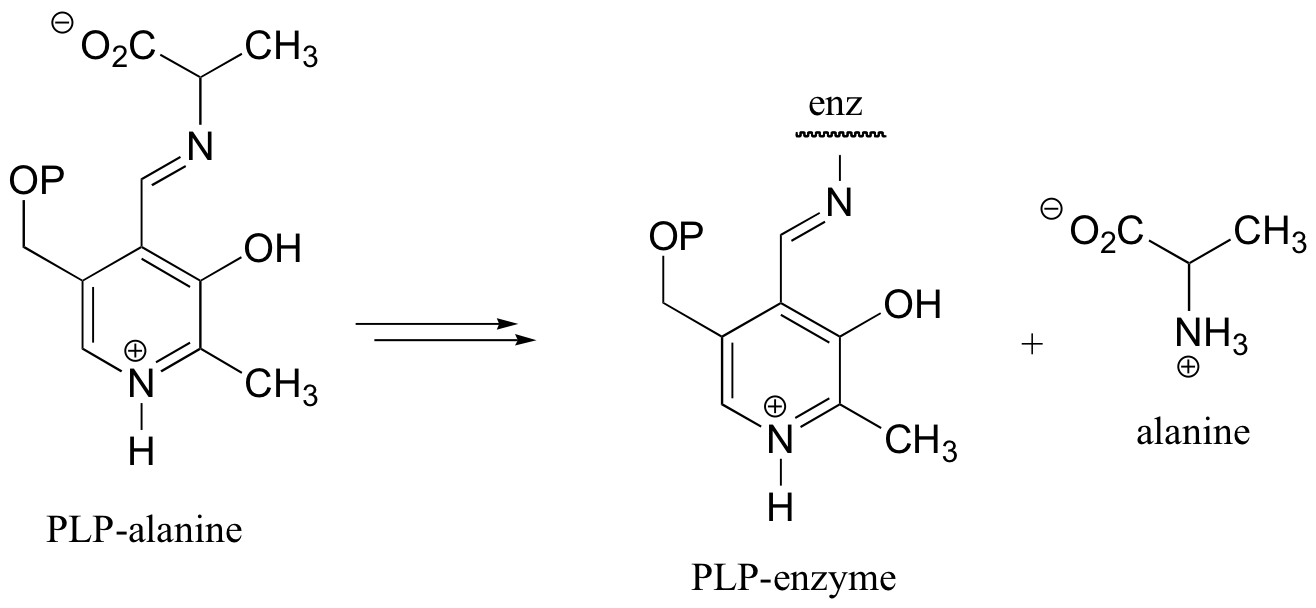
c)
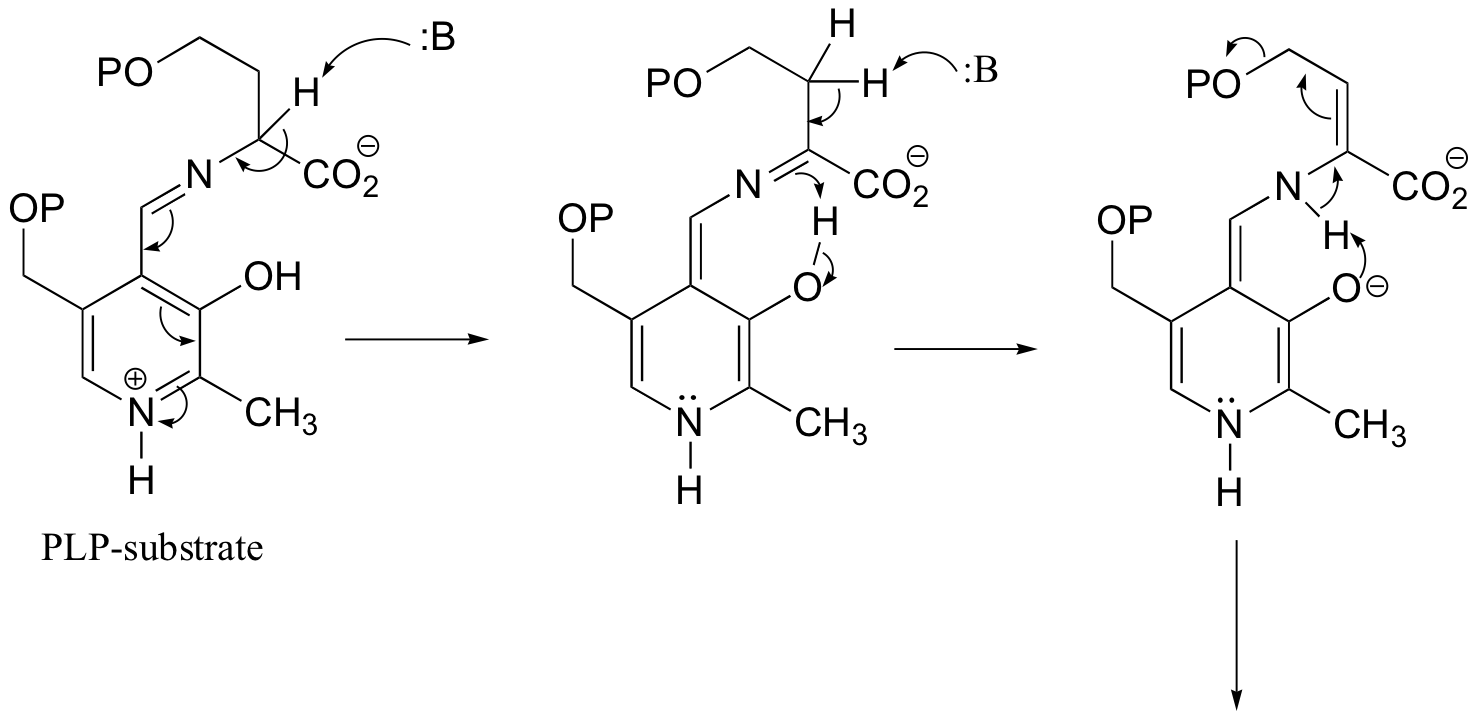
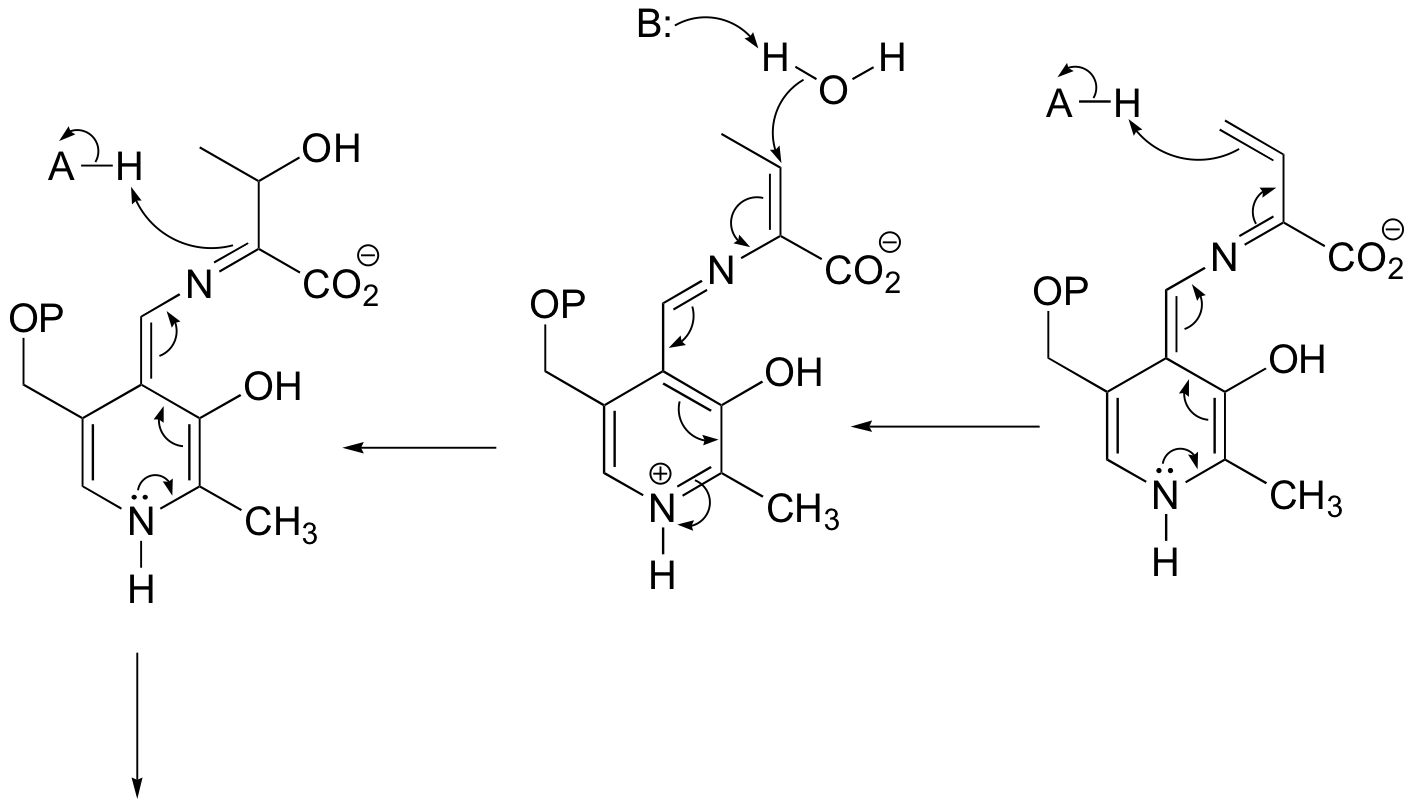
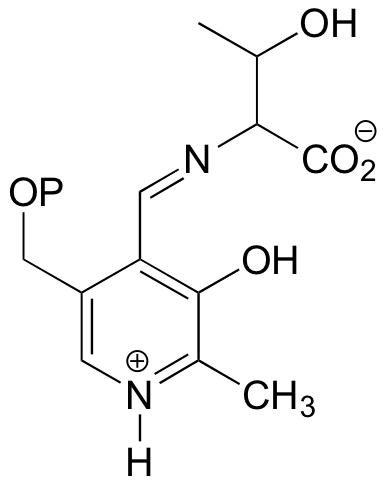
P14.11:
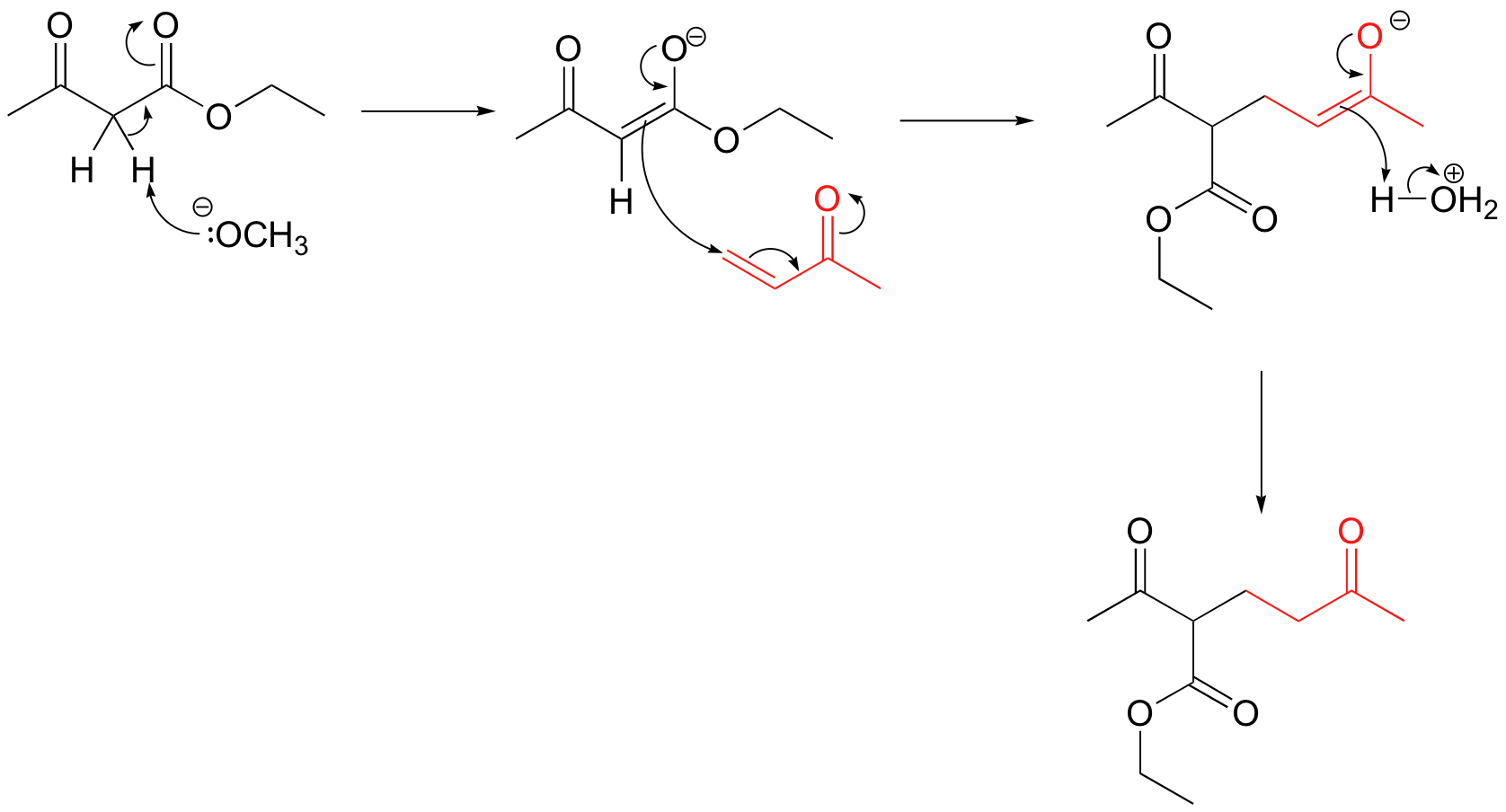
P14.12:
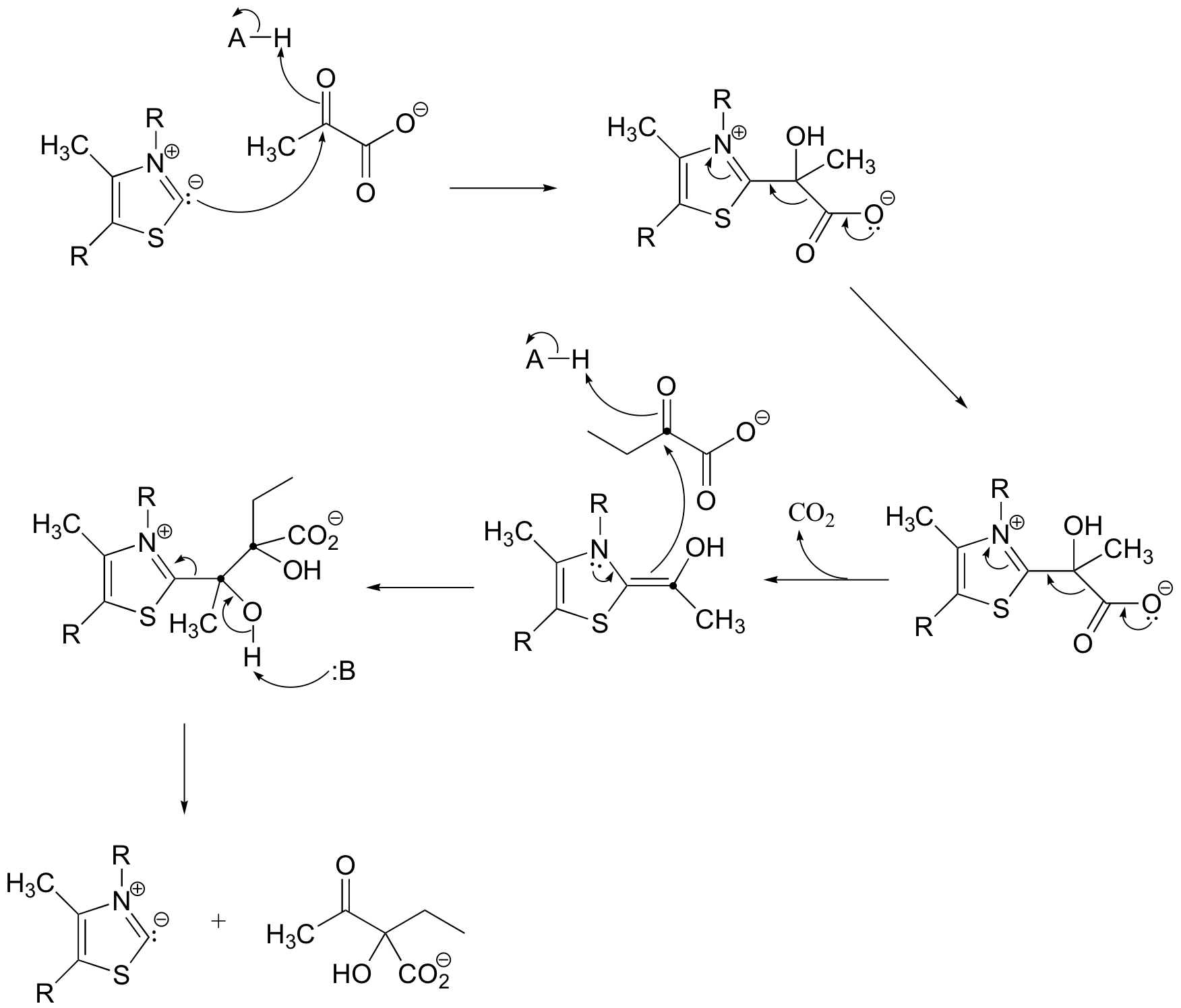
P14.13:
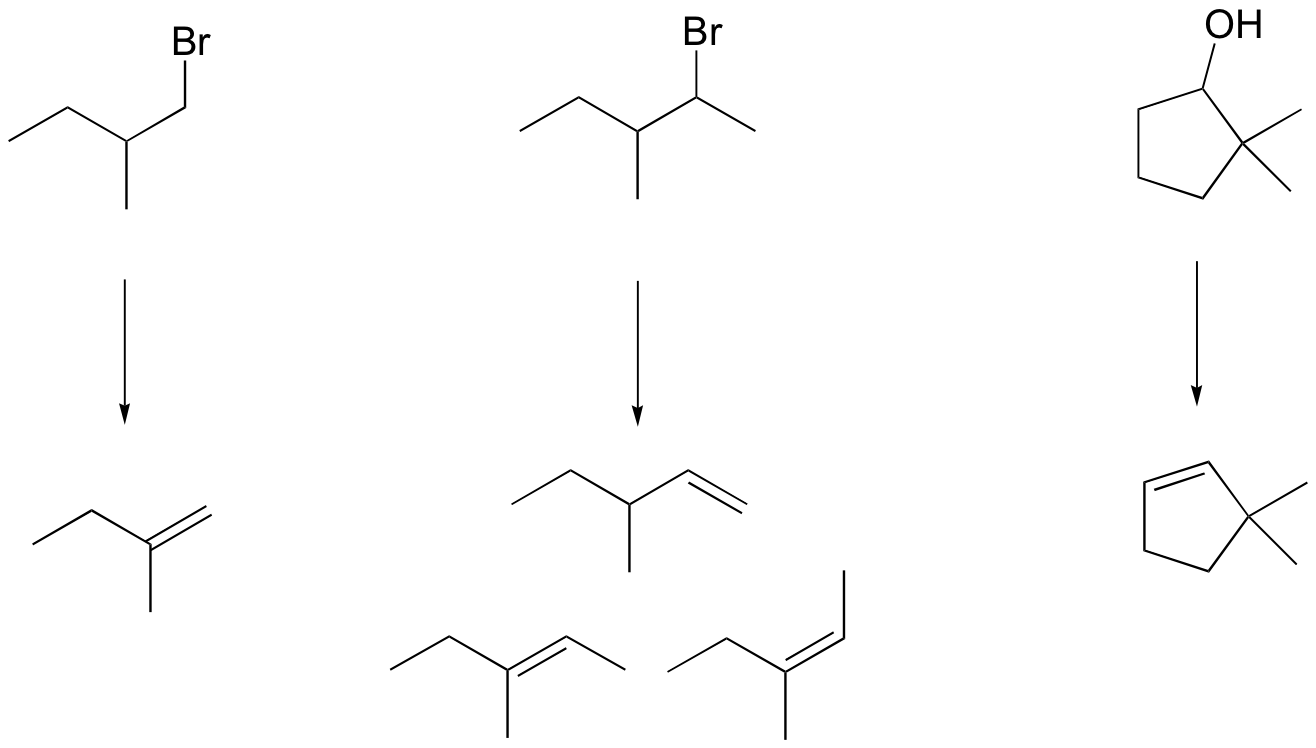
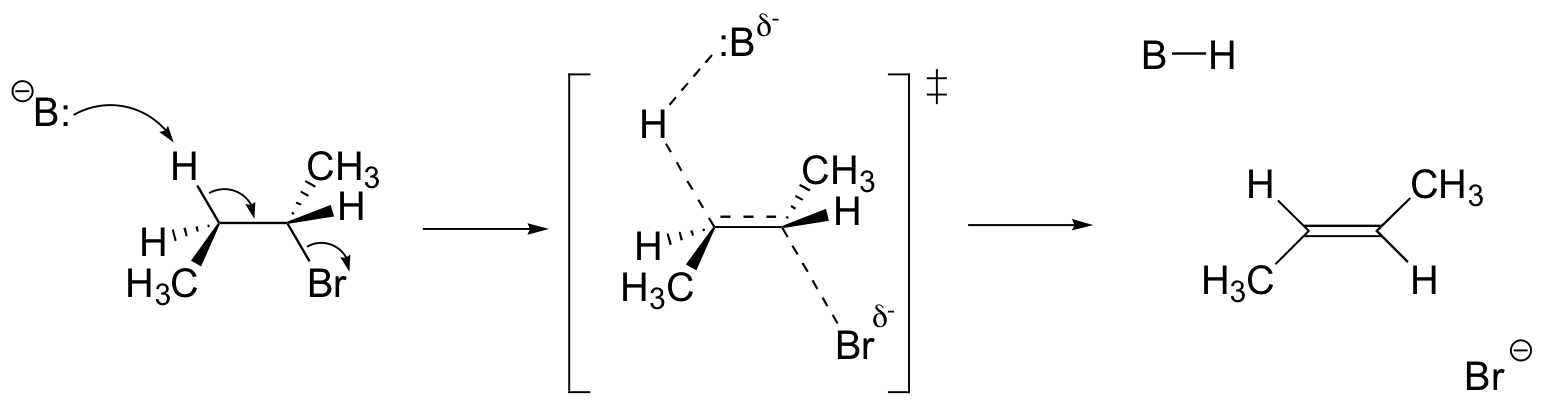
P14.14: Notice that in order for the (E)-alkene to form, the C-H and C-Br bonds must be anti and co-planar.
P14.15: Recall that for an E2 reaction to occur on a cyclohexane ring, the leaving group and the abstracted hydrogen must both be in axial positions.

P14.16:
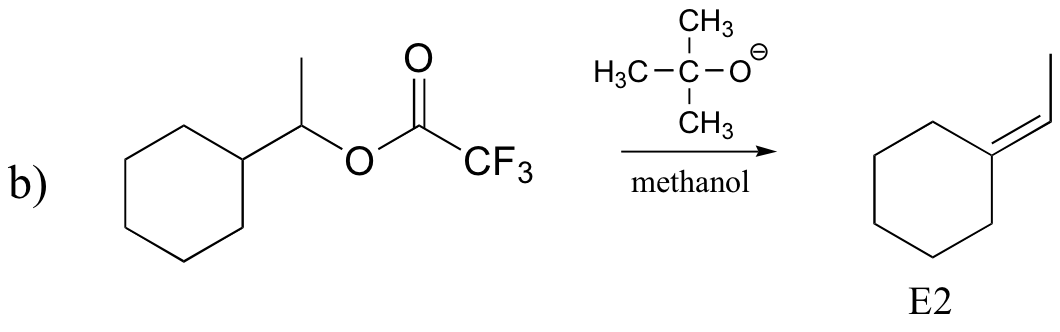
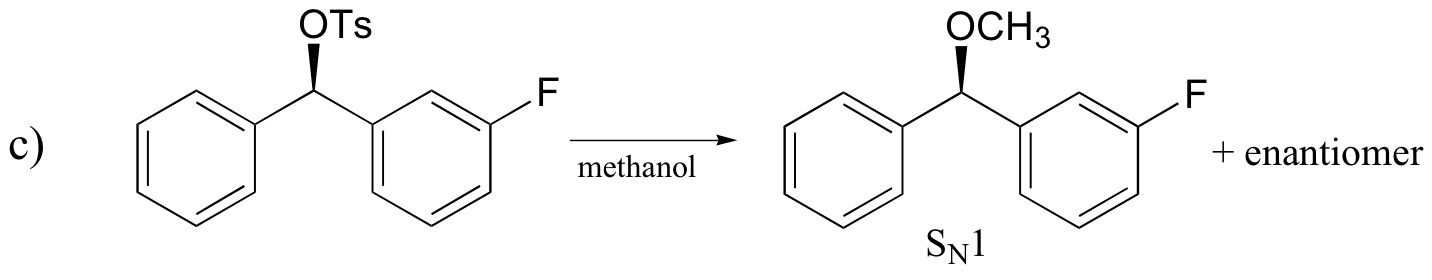
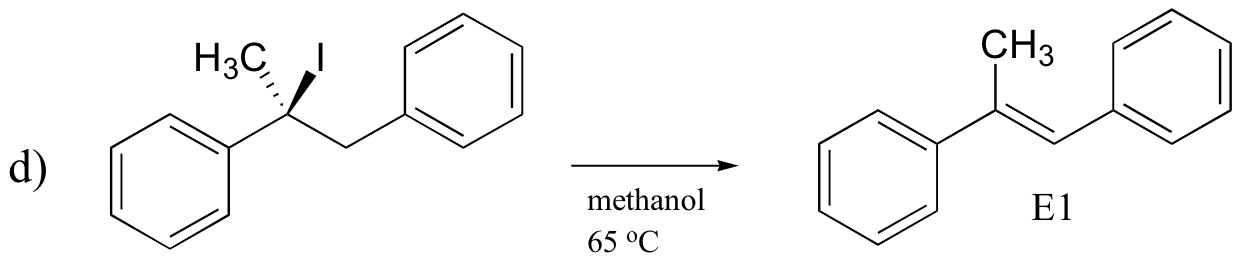
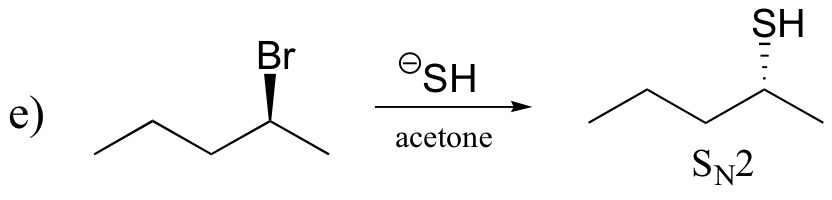
P14.17:
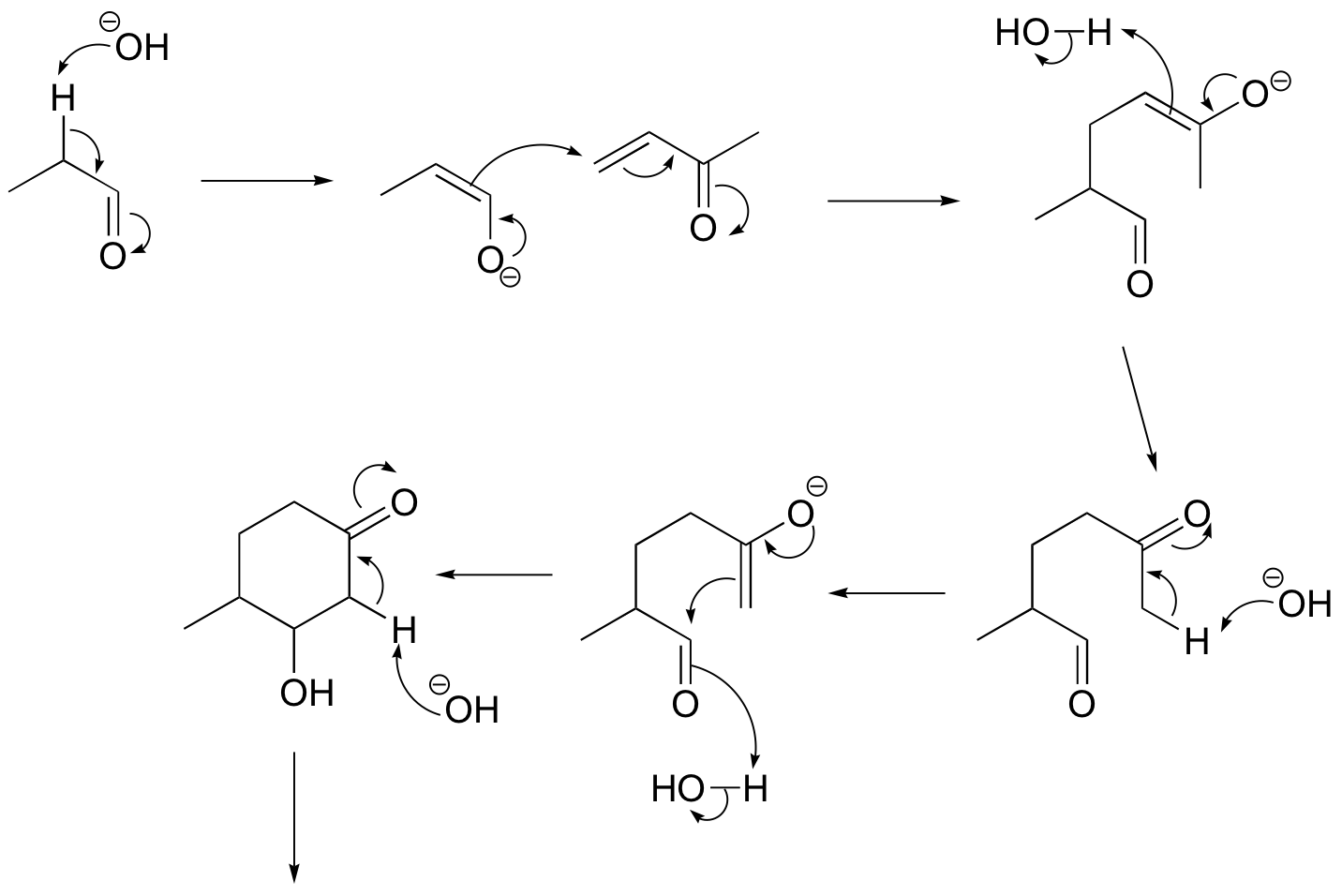

P14.18:
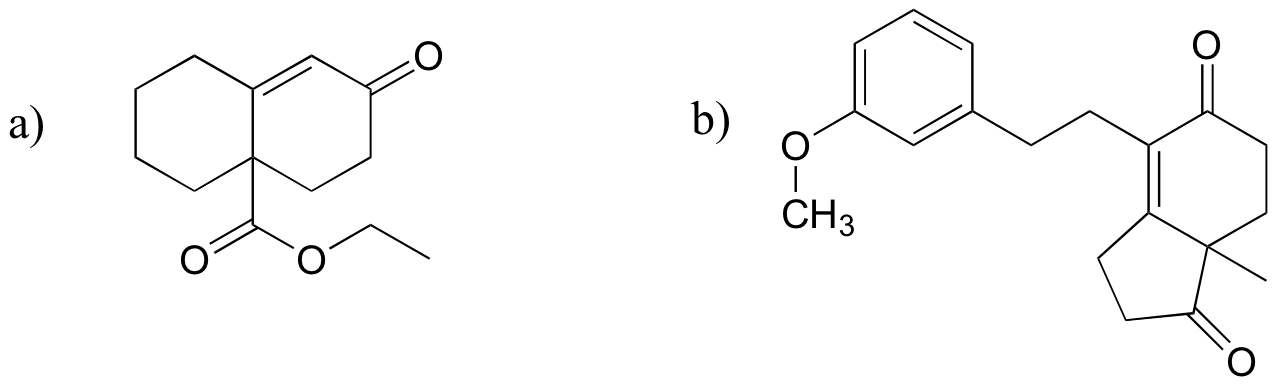
P14.19:
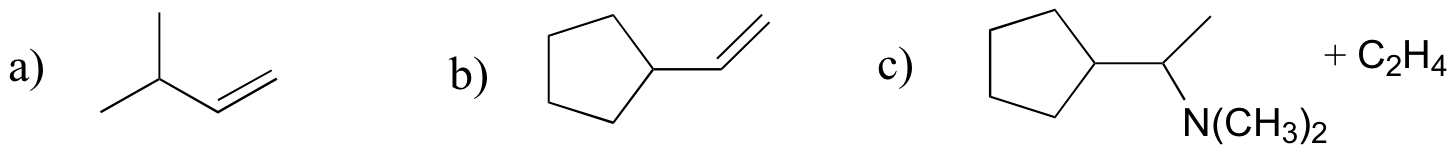
a)
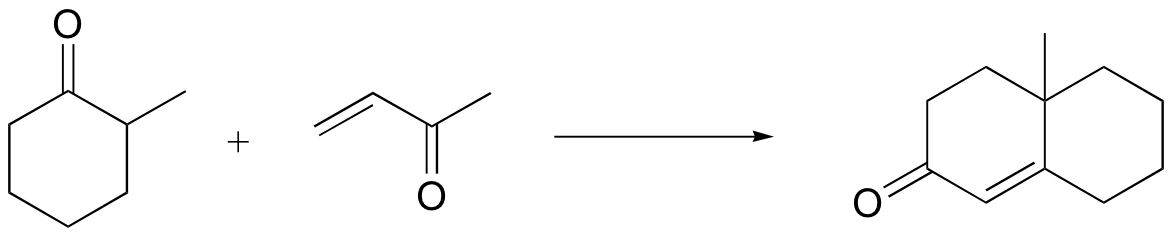
b)
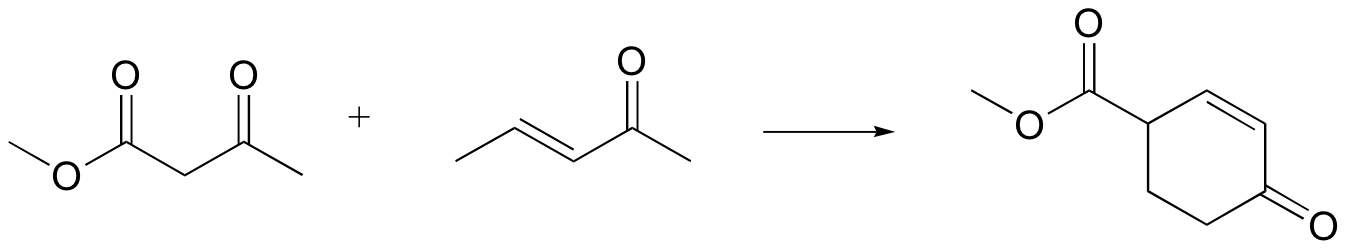
P14.20:
Challenge problems
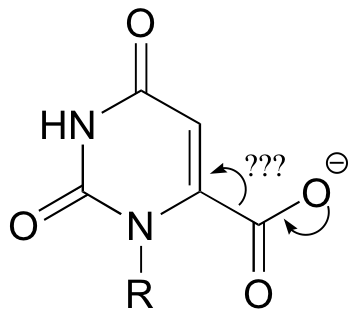
C14.1:
a) It is a decarboxylation reaction, but there is no obvious way for the electron pair to be stabilized.
b) A carbene intermediate would imply a very hydrophobic active site pocket – see Chemical and Engineering News, May 12, 1997, p. 12; Science 1997, 276, 942.
c) See Chemical and Engineering News, March 13, 2000, p. 42.
C14.2: See Biochemistry 2003, 42, 248 (scheme 1)
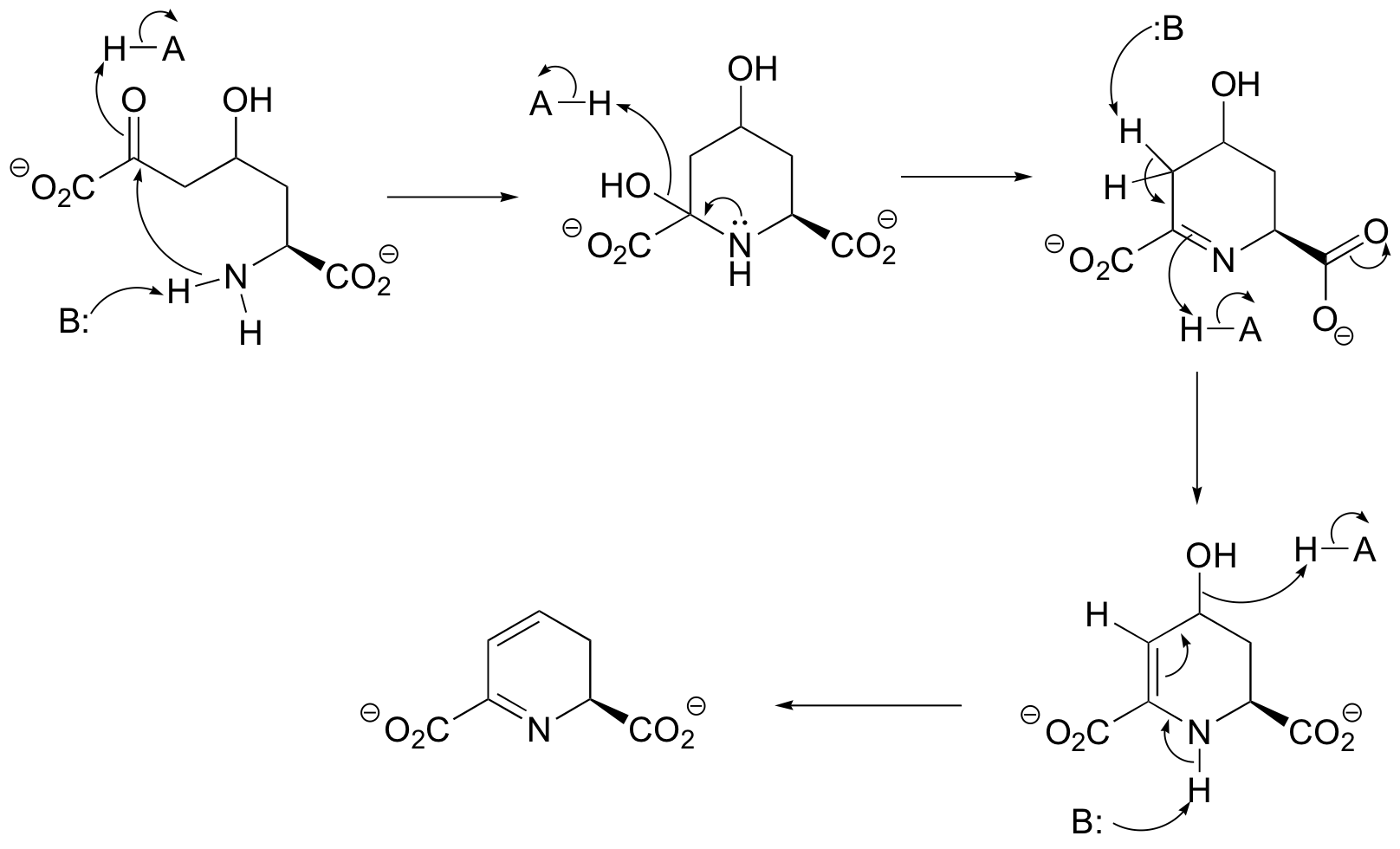
C14.3: The first part is cyclic imine formation, then we see an E1cb dehydration adjacent to the imine.
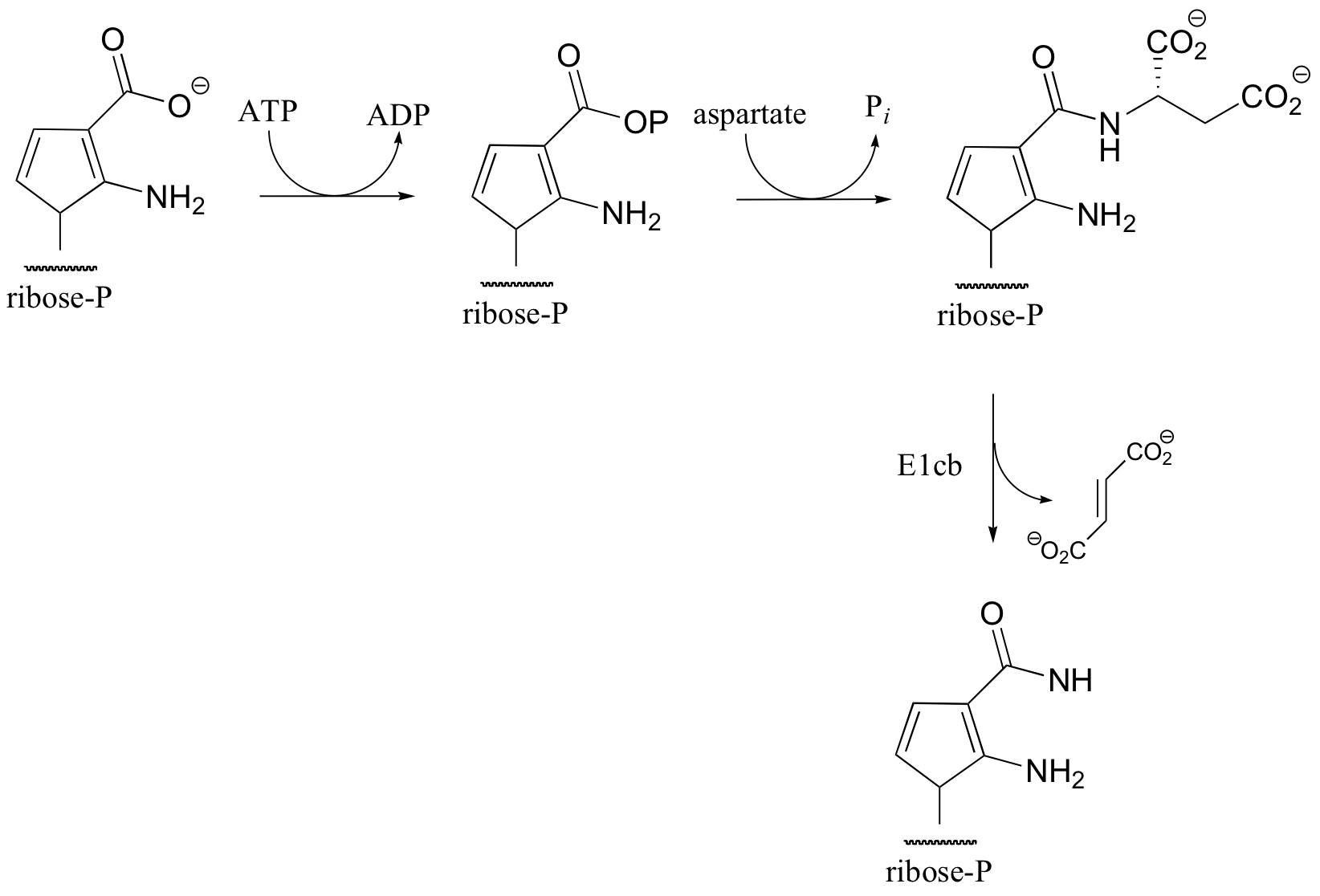
C14.4:
a) Lacking any other information, it is reasonable to expect a mechanism that parallels that of asparagine synthetase (section12.2B): conversion to an acyl phosphate, then acyl transfer with ammonia nucleophile.
b) The nitrogen comes from the amino group on aspartate, and is freed by the final elimination step.
C14.5: See European J. Biochem. 2002, 269, 1790, fig 7.