
Chapter 8 Solutions
- Page ID
- 1123

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In-chapter exercises
E8.1:
E8.2:
Notice that the reaction occurs with inversion of configuration: the leaving group (I) was pointing out of the plane of the page, while the nucleophile (CH3S-) attacks from behind, and ends up pointing into the plane of the page.
E8.3:
The intermediate species is a carbocation that forms after the bromine leaves:

E8.4:
E8.5: Reaction A is a concerted (one-step) reaction, essentially a collision between two species. Therefore, if the concentration of nucleophile (CH3S-) were doubled in the solution, the rate of the reaction should also double. You should recall learning about rate expressions in your general chemistry course - this is an example of a second order rate expression, where the rate depends on some rate constant (k) and the product of two concentration:
rate = k[CH3I] [CH3S-]
(If this is unfamiliar to you, now would be a good time for a quick review of rate expressions in your general chemistry textbook!)
Reaction B is a two-step reaction. The first step – the breaking of the C-Br bond – is the slow, rate determining step, and does not involve the CH3SH nucleophile at all. Therefore, changing the concentration of the nucleophile should have no effect on the rate of the reaction. (If the concentration of CH3SH were doubled, the second step would occur twice as fast – but the rate of the first, slower step would not change, and so the overall rate of the reaction would not be affected). The rate expression in this case is first order (it depends on the concentration of only one reactant):
rate = k[C(CH3)3Br]
E8.6:
E8.7: The side chain on serine – a primary alcohol – is in general a better nucleophile then the side chain on tyrosine, which is a phenol. Recall (section 7.4A) the argument for why phenolate (deprotonated phenol) is less basic than ethanoate (deprotonated ethanol) – some of the electron density of the oxygen atom in phenolate is delocalized to the aromatic ring. The same argument holds true when comparing the nucleophilicity of serine to tyrosine: electron density on the tyrosine oxygen is stabilized somewhat by resonance with the benzene ring, and is therefore less reactive. Steric factors also come into play - the large phenyl ring makes nucleophilic attack more difficult for the tyrosine.
E8.8: A cysteine, with its thiol side chain, is very nucleophilic - much more so than a methionine with its sulfide group. The thiol sulfur atom is less hindered than the sulfur atom on a sulfide, and in addition the thiol proton can be removed by a base as the nucleophilic attack takes place. The methyl group on a methionine side chain, in contrast, cannot be removed by a base, so that the product of a nucleophilic attack by methionine will contain a positive formal charge on the sulfur.
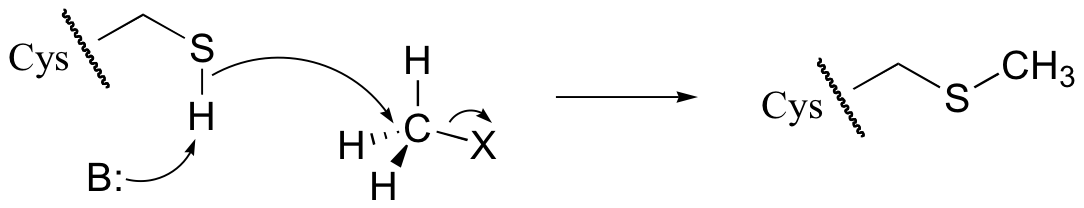
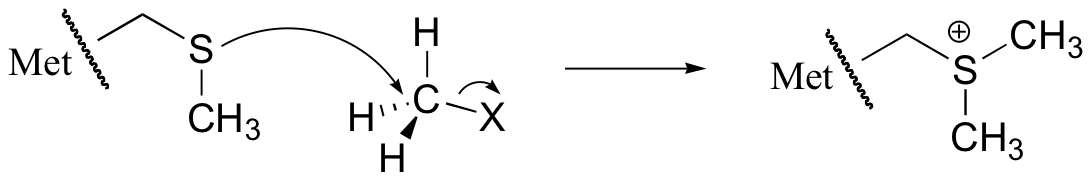
E8.9:
a) phenolate is the stronger base (compare the pKas of the conjugate acids), and is also the stronger nucleophile – the electrons on the nucleophilic oxygen atoms are more reactive.
b) water is nucleophilic, hydronium ion is not – the difference is in the protonation state (deprotonation increases nucleophilicity)
c) trimethyl amine is less hindered, and therefore is the better nucleophile.
d) in a polar aprotic solvent like acetone, chloride will be the stronger nucleophile.
e) CH3NH- is anionic and deprotonated – it is the stronger nucleophile.
E8.10:
In the carbocation on the left, the positive charge is located in a position relative to the nitrogen such that the lone pair of electrons on the nitrogen can be donated to fill the empty orbital. This is not possible for the carbocation species on the right.
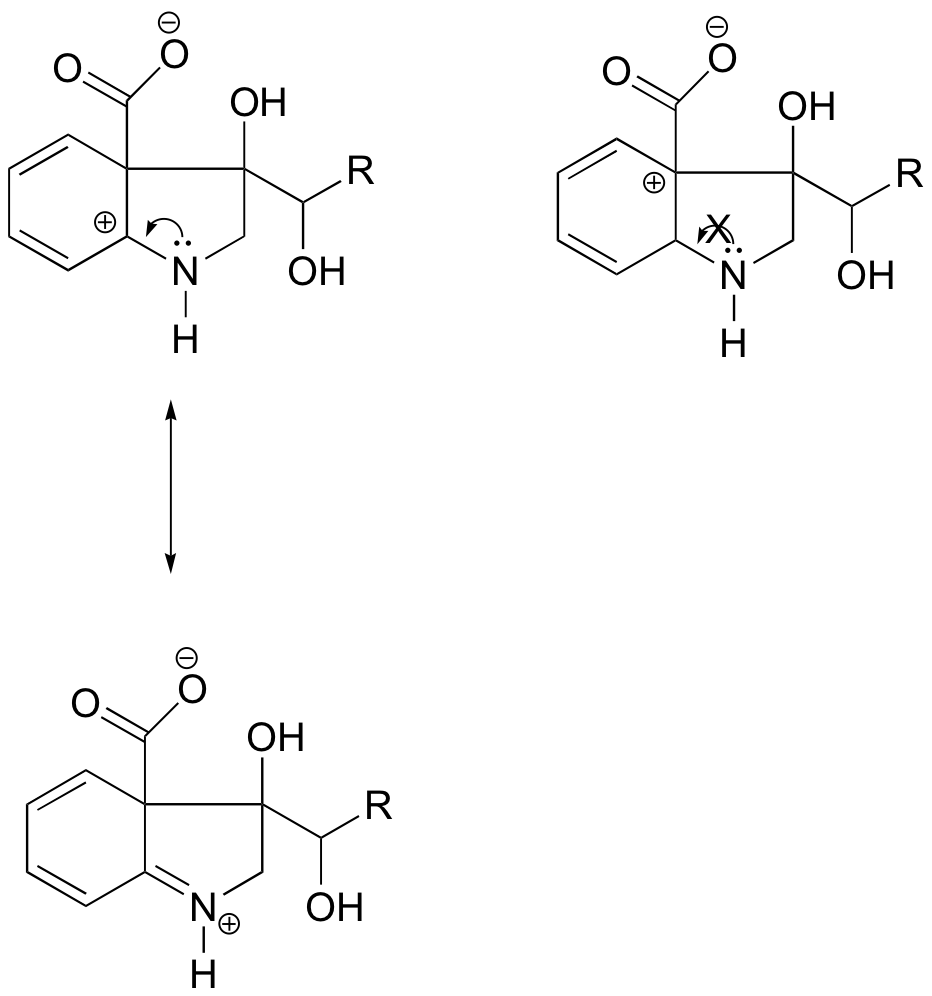
E8.11:
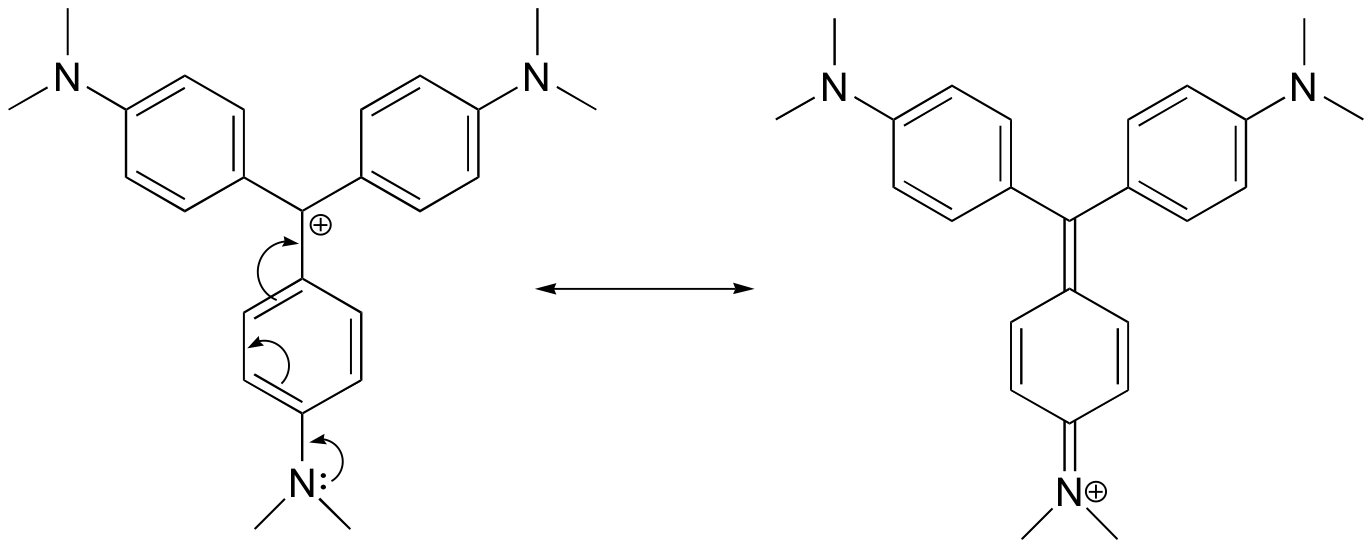
E8.12:
a) 1 (tertiary vs. secondary carbocation)
b) equal
c) 1 (tertiary vs. secondary carbocation)
d) 2 (positive charge is further from electron-withdrawing fluorine)
e) 1 (lone pair on nitrogen can donate electrons by resonance)
f) 1 (allylic carbocation – positive charge can be delocalized to a second carbon)
E8.13:
a) B (thiolate is a weaker base/better leaving group than alcohol)
b) A (sulfide is better leaving group than thiolate (sulfide is neutral and not at all basic)
c) A (bromide ion is a weaker base/better leaving group than acetate – compare pKa values of HBr and acetic acid).
d) B (inductive electron-withdrawing effect of fluorines make trifluoroacetate a weaker base/better leaving group).
E8.14:
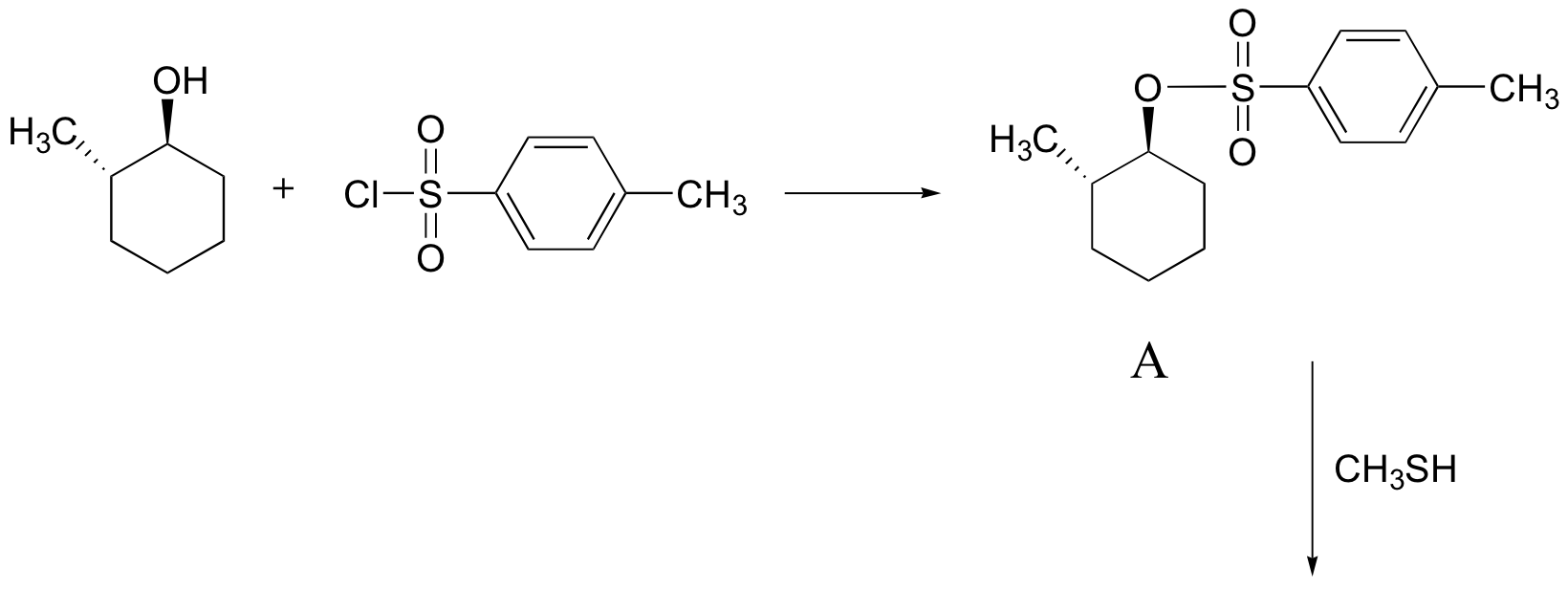
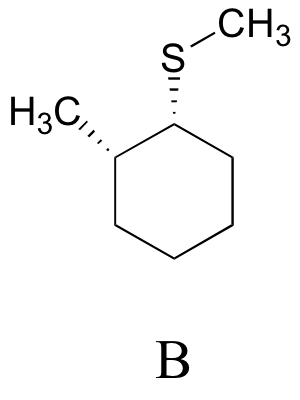
E8.15:
a) SN2 (primary electrophile, strong nucleophile, polar aprotic solvent)
b) SN1 (tertiary electrophile, weak nucleophile, protic solvent)
c) SN2 (secondary electrophile, strong nucleophile, polar protic solvent)
E8.16:
a) Notice that the oxygen nucleophile attacks from behind the plane of the page, while the chlorine is pointing out of the plane of the page – this is a backside attack by the nucleophile.
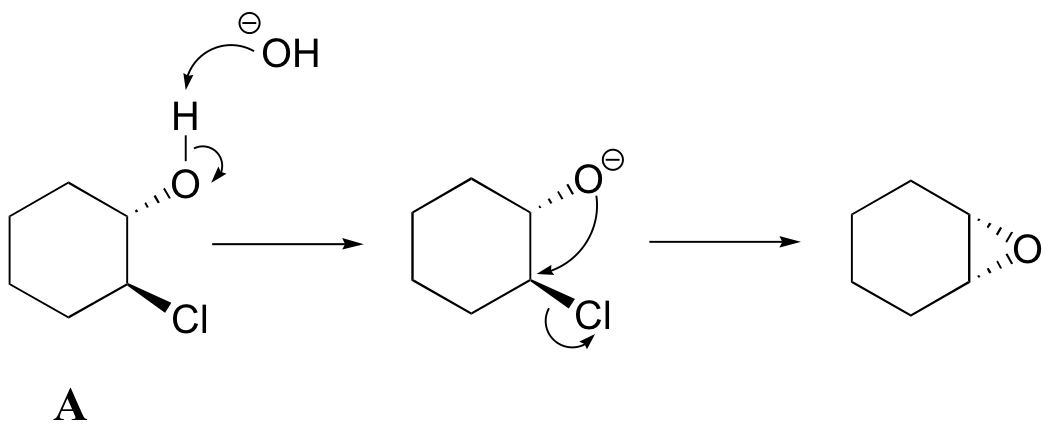
b) Because both the nucleophilic oxygen and the chlorine leaving group are oriented on the same side of the ring, backside attack by the nucleophile is impossible. In order for a displacement to occur, it would have to be a stepwise (SN1) mechanism. The observation that compound A forms an epoxide under these conditions but compound B does not strongly suggests that the reaction proceeds by an SN2 mechanism, with a requirement for backside attack by the nucleophile.
E8.17:
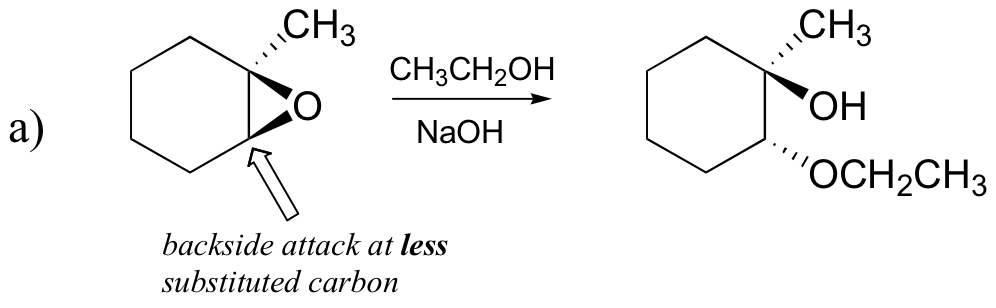
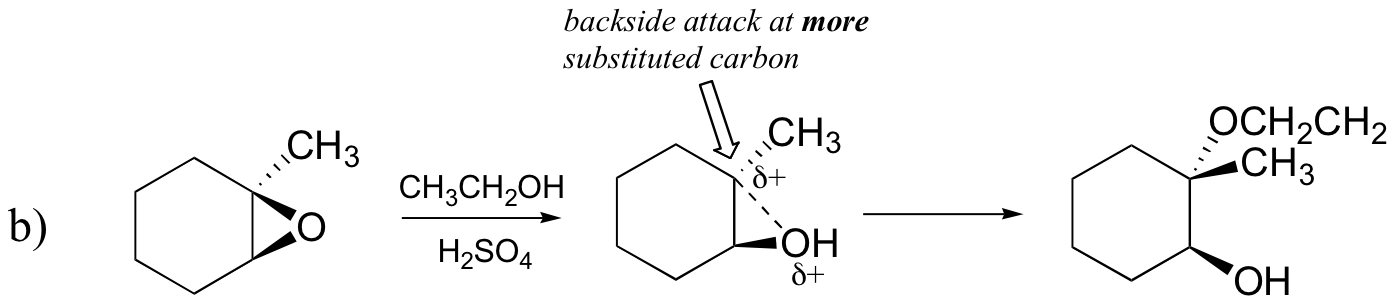
End-of-chapter problems
P8.1: All of the molecules in question are primary alkyl bromides, and the nucleophile is a very powerful thiolate ion – we are talking here about SN2 reactions. The rate of substitution depends on the amount of steric hindrance in the electrophile – the less hindrance, the faster the substitution reaction. The order is:
fastest D > B > A > C > slowest
P8.2:
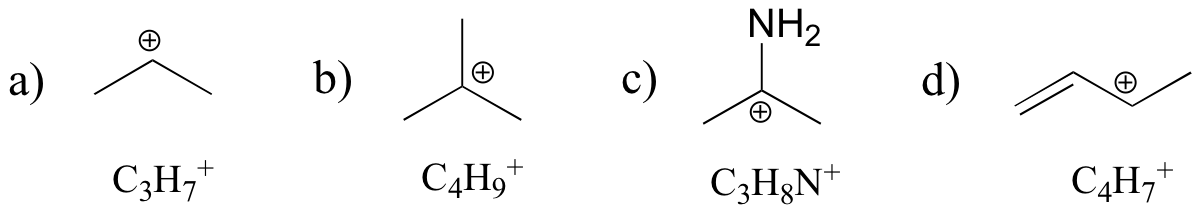
P8.3:
Carbocation A is more stable: it has a longer system of conjugated p bonds, and as a consequence the charge can be delocalized over five different carbons (as opposed to four carbons for B).
P8.4:
Here we are looking at periodic trends and steric hindrance. Nucleophilicity increases going down a column of the periodic table, so A and C, with phosphorus atoms, are expected to be more nucleophilic than B and D. C is more nucleophilic than A, because the three methyl groups on C are the cause of less steric hindrance than the three ethyl groups on A. Using the same reasoning, we can see that B should be more nucleophilic than D, because of the bulky phenyl group on D. The trend is:
most nucleophilic C > A > B > D least nucleophilic
P8.5:
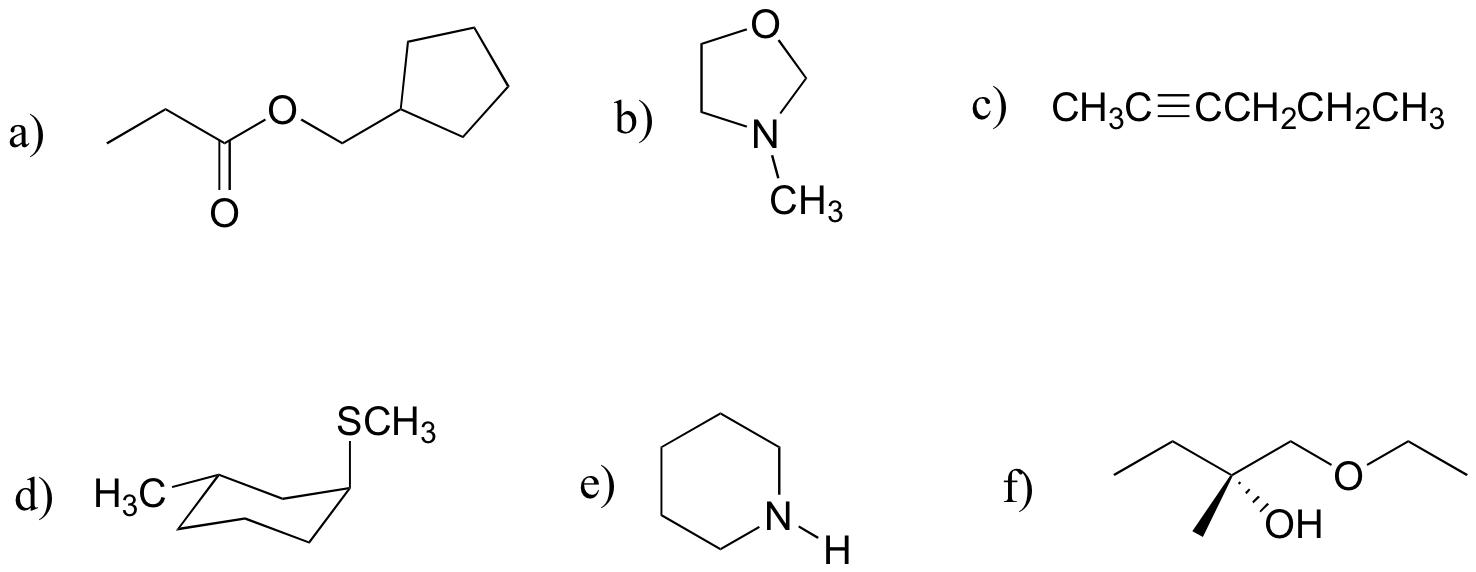
P8.6:
The reaction shown in part e) is intramolecular (one molecule reacting with itself), and thus will likely have a first order rate expression.
P8.7:
a) water or hydroxide ion
b) CH3S- or CH3OH
c) CH2S- or CH3SH
d) acetate ion or hydroxide ion
e) diethyl sulfide or diethyl ether
f) dimethylamineor diethylether
g) trimethylamine or 2,2-dimethylpropane
P8.8: The major product will be dimethyl sulfide (CH3SCH3), because CH3S- is a better nucleophile than CH3O- and will react faster with methyl bromide in an SN2displacement.
P8.9:
a) the compound on the left
b) the compound on the right
c) the compound on the right
d) the compound on the left
e) the compound on the left
f) the compound on the left
g) the compound on the right
P8.10:

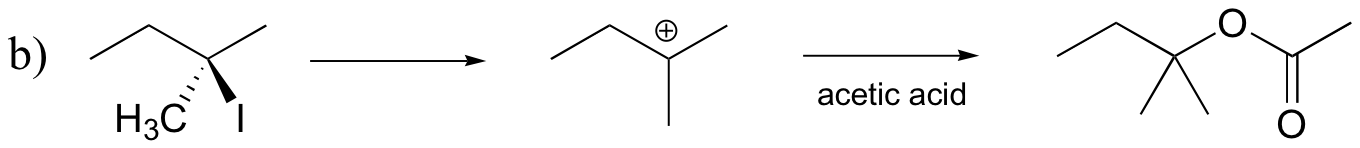
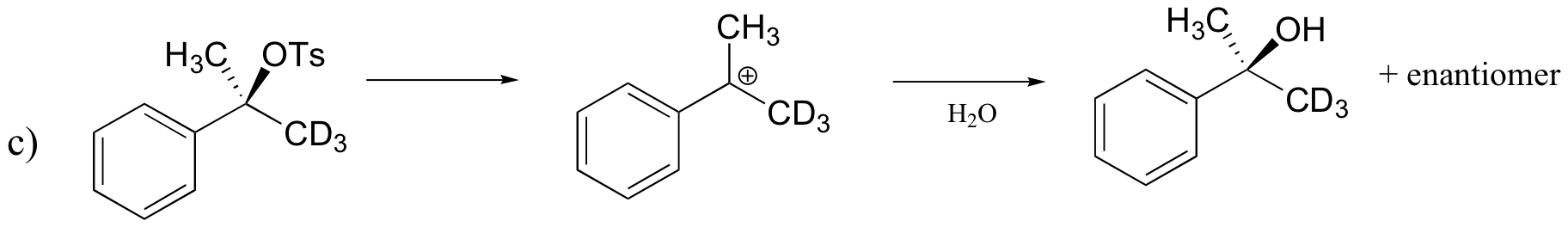
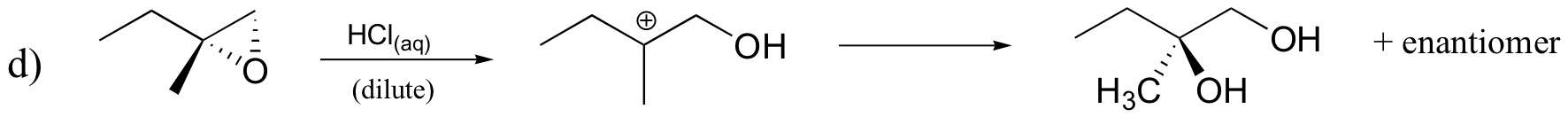
P8.11:
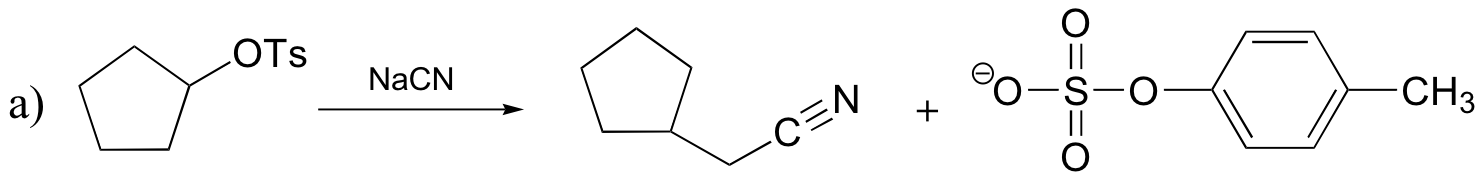
![]()
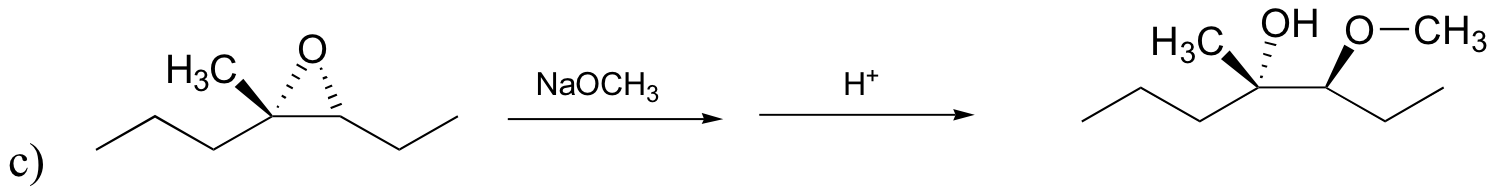
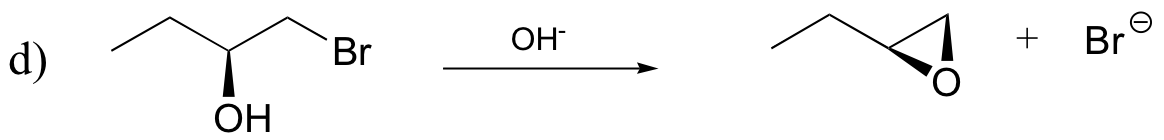
P8.12:
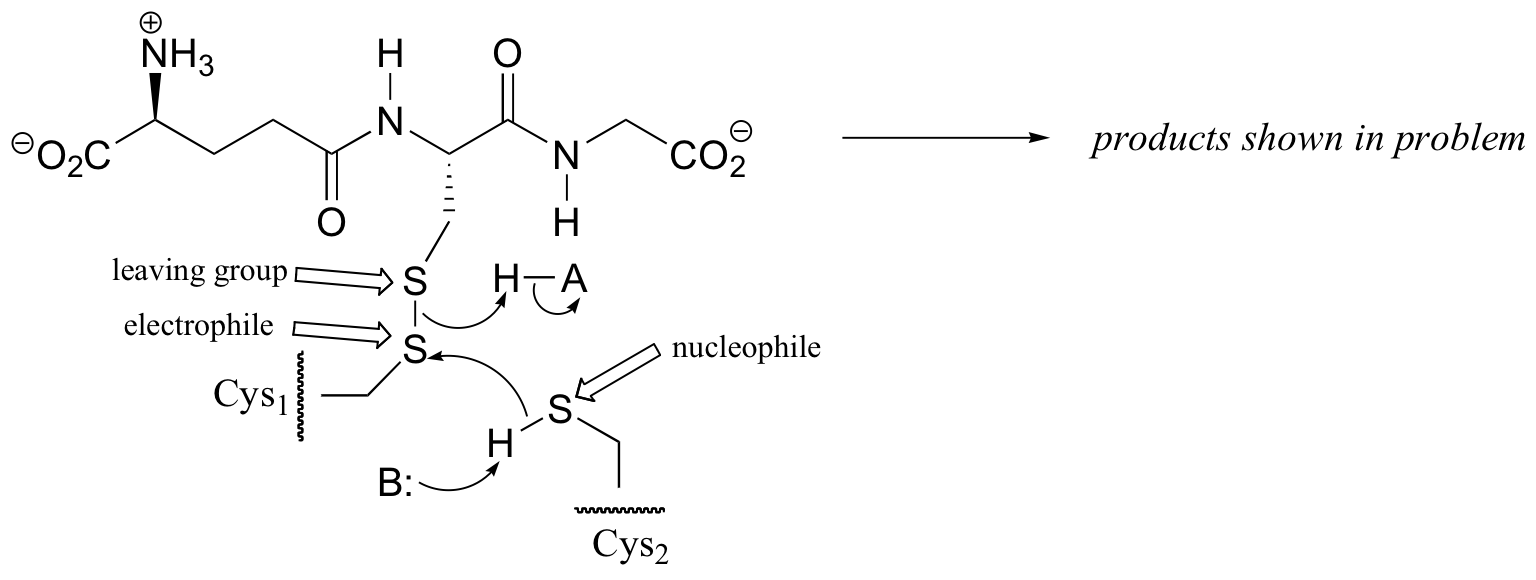
P8.13:
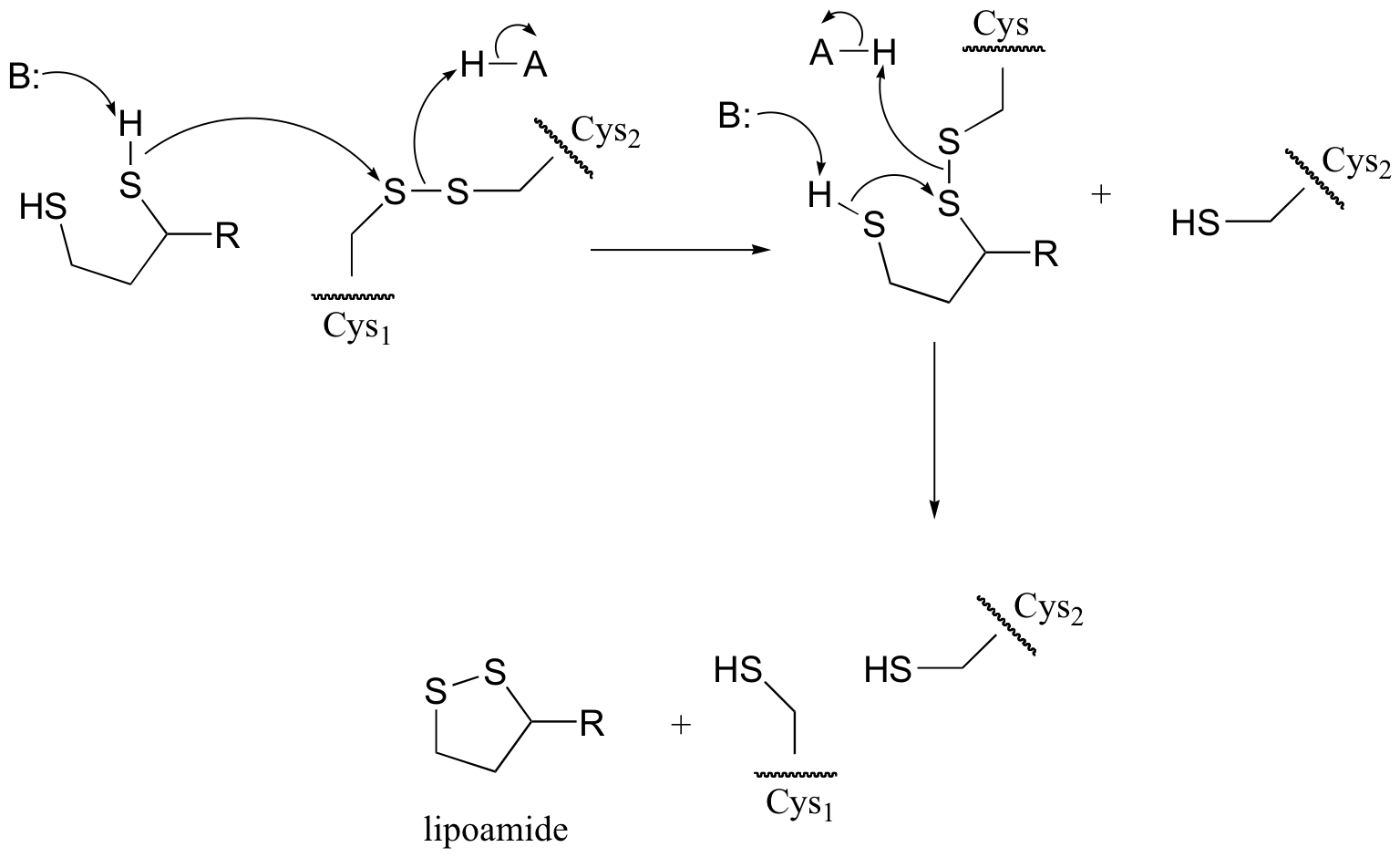
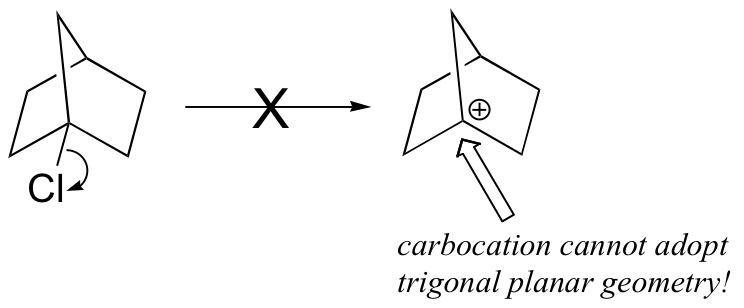
P8.14:
The first step in an SN1 reaction is carbocation formation. However, the rigid ‘bicyclo’ structure of the starting material prevents a hypothetical carbocation intermediate from adopting trigonal planar geometry (make a model to see this better). Consequently, there is a large energy barrier for carbocation formation.
Challenge problems
C8.11:
See the following references for a solution:
March, Jerry, Advanced Organic Chemistry: Reactions and Mechanisms, and Structure, 4th ed. (1992), p. 296. (Use the 'author index' to find a discussion of this paper in more recent editions of the book).
J. Chem Soc. 1935, 1525. (This is the original paper, but may be hard to locate.)
Contributors
- Organic Chemistry With a Biological Emphasis, Tim Soderberg (University of Minnesota, Morris)