
16.4: Hydrogenation/dehydrogenation reactions of carbonyls, imines, and alcohols
- Page ID
- 1012

Now that we have a good handle on the big picture of reduction and oxidation, let's look at the actual chemical mechanisms involved in the hydrogenation (reduction) and dehydrogenation (oxidation) of organic molecules. We will concentrate first on hydrogenation and dehydrogenation reactions involving aldehydes, ketones, imines (Schiff bases), and carboxylic acid derivatives. Hydrogenation (reduction) of an aldehyde, ketone, or imine results in a primary alcohol, secondary alcohol, and amine, respectively. Mechanistically, these reactions are variations on the chapter 11 reactions: they are simply nucleophilic additions to a carbonyl or imine, with a hydride ion acting as the nucleophile.
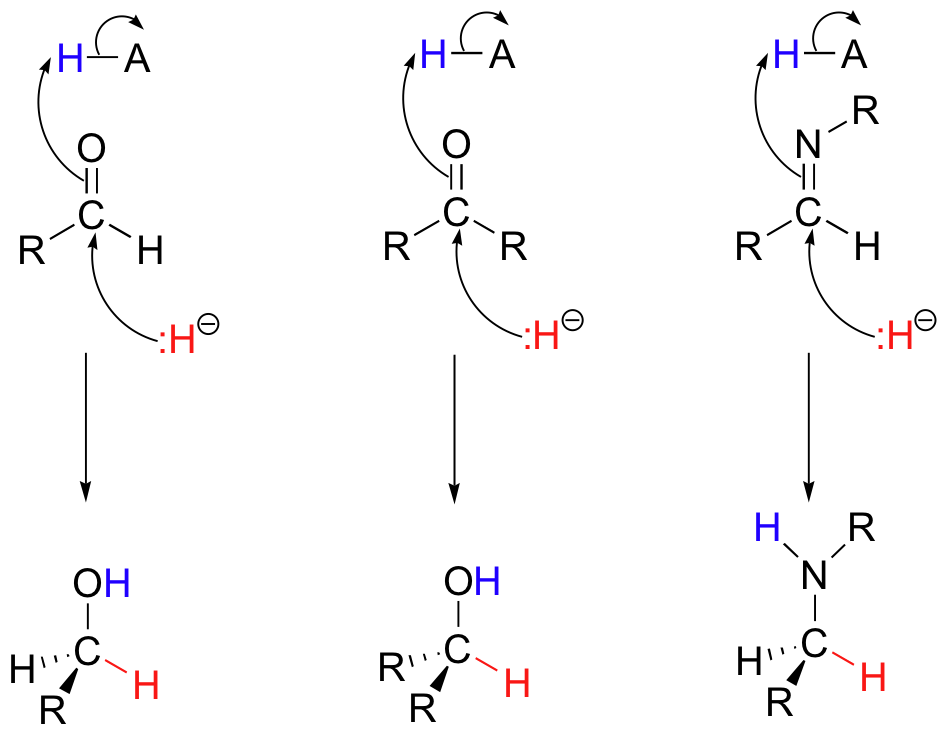
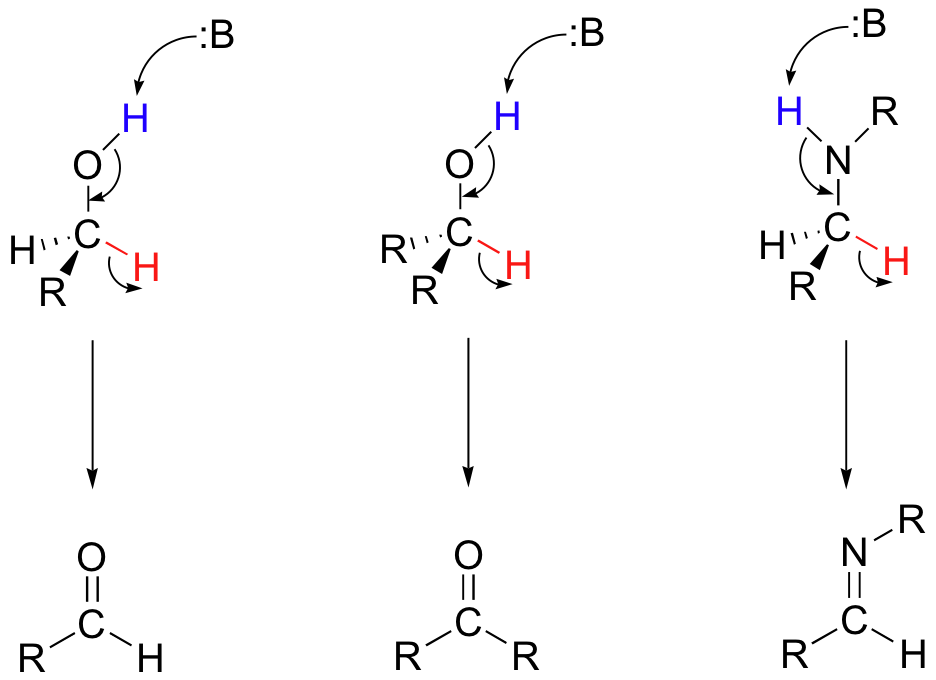
In the reverse direction, dehydrogenation (oxidation) of a primary alcohol, secondary alcohol, or amine results in an aldehyde, ketone, or imine, respectively.
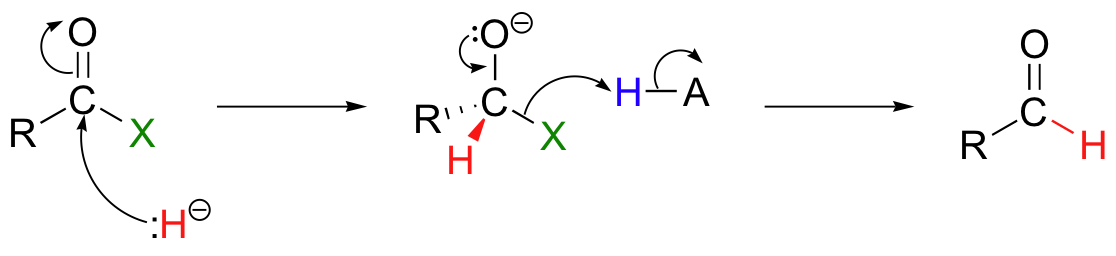
Hydrogenation of a carboxylic acid derivative results in an aldehyde. Mechanistically, this is a nucleophilic acyl substitution with a hydride ion acting as the nucleophile:
Dehydrogenation of an aldehyde results in a carboxylic acid derivative: again, this is simply a nucleophilic acyl substitution, except this time the hydride is the leaving group:
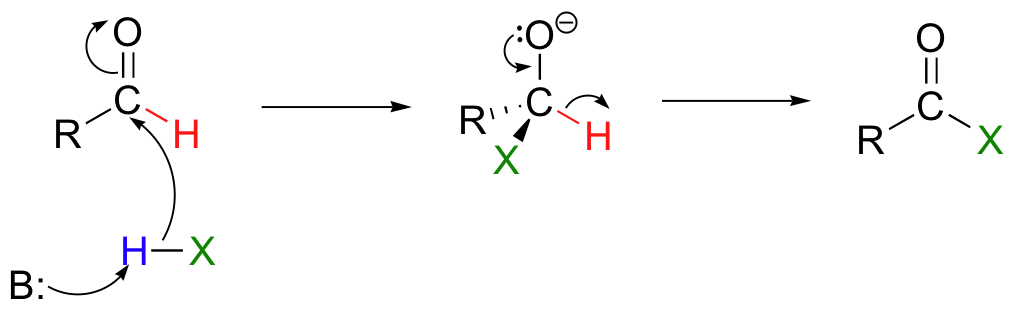
Both of these reactions can be described as hydride transfer reactions.
16.4A: Nicotinamide adenine dinucleotide - a hydride transfer coenzyme
Although we are talking in this chapter about hydrides acting as nucleophiles and leaving groups, you already know that discreet hydride ions are far too unstable to exist as actual intermediates in the organic reactions of living cells. Biochemical redox reactions involving hydride transfer require the participation of a hydride transfer coenzyme. In reactions involving carbonyl compounds, a molecule called nicotinamide adenine dinucleotide generally plays this role. The full structure of the oxidized form of this coenzyme, abbreviated NAD+, is shown below, with the active nicotinamide group colored blue.
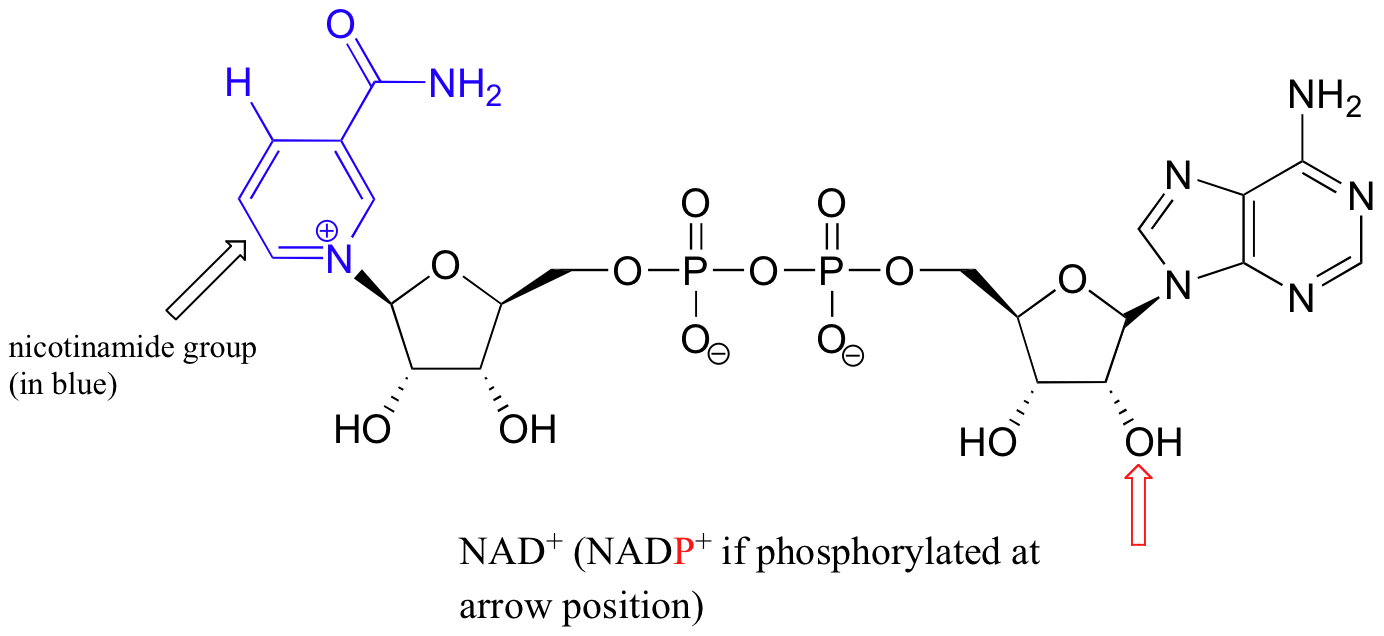
If the hydroxyl group indicated by the arrow is phosphorylated, the coenzyme is called NADP+. Because the redox chemistry occurs specifically at the nicotinamide part of the molecule, typically the rest of the molecule is simply designated as an 'R' group.
NAD+ and NADP+ both function in biochemical redox reactions as hydride acceptors: that is, as oxidizing agents. The other forms of the coenzyme, NADH and NADPH, serve as hydride donors: that is, as reducing agents.
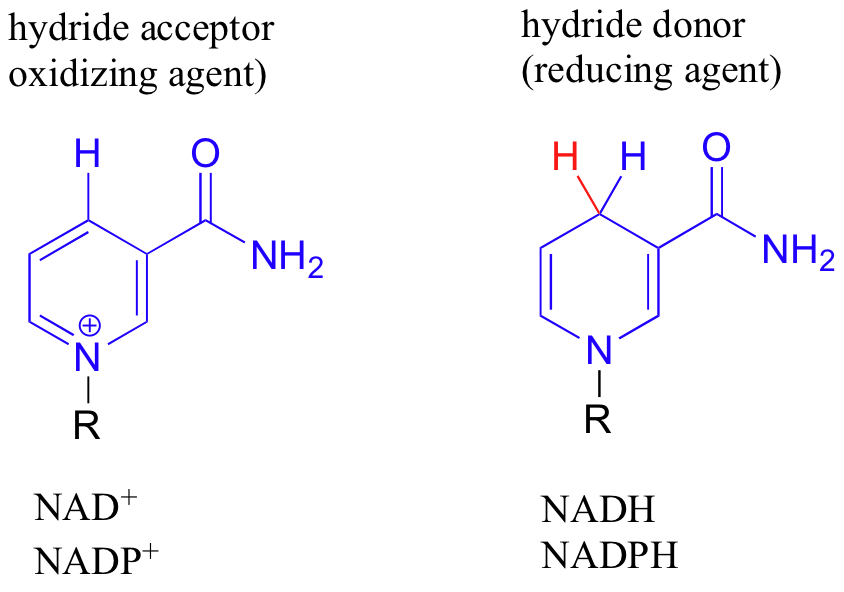
The phosphate on the nucleotide pentose group of NADP+ and NADPH is located far from the nicotinamide ring, and does not participate directly in the hydride transfer function of the cofactor. It is, however, important in a larger metabolic context: as a general rule, redox enzymes involved in catabolism (the breakdown of large molecules) typically use NAD+ and NADH, while those involved in anabolism (synthesis of large molecules from small precursors) use the phosphorylated coenzyme (NADP+ and NADPH).
16.4B: Carbonyl hydrogenation and alcohol dehydrogenation - the general picture
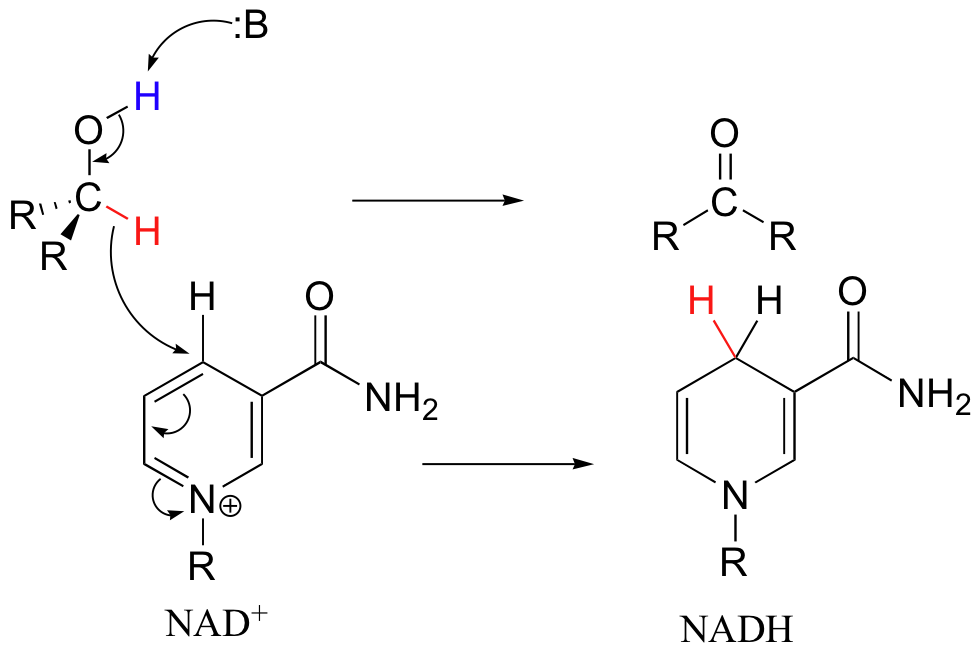
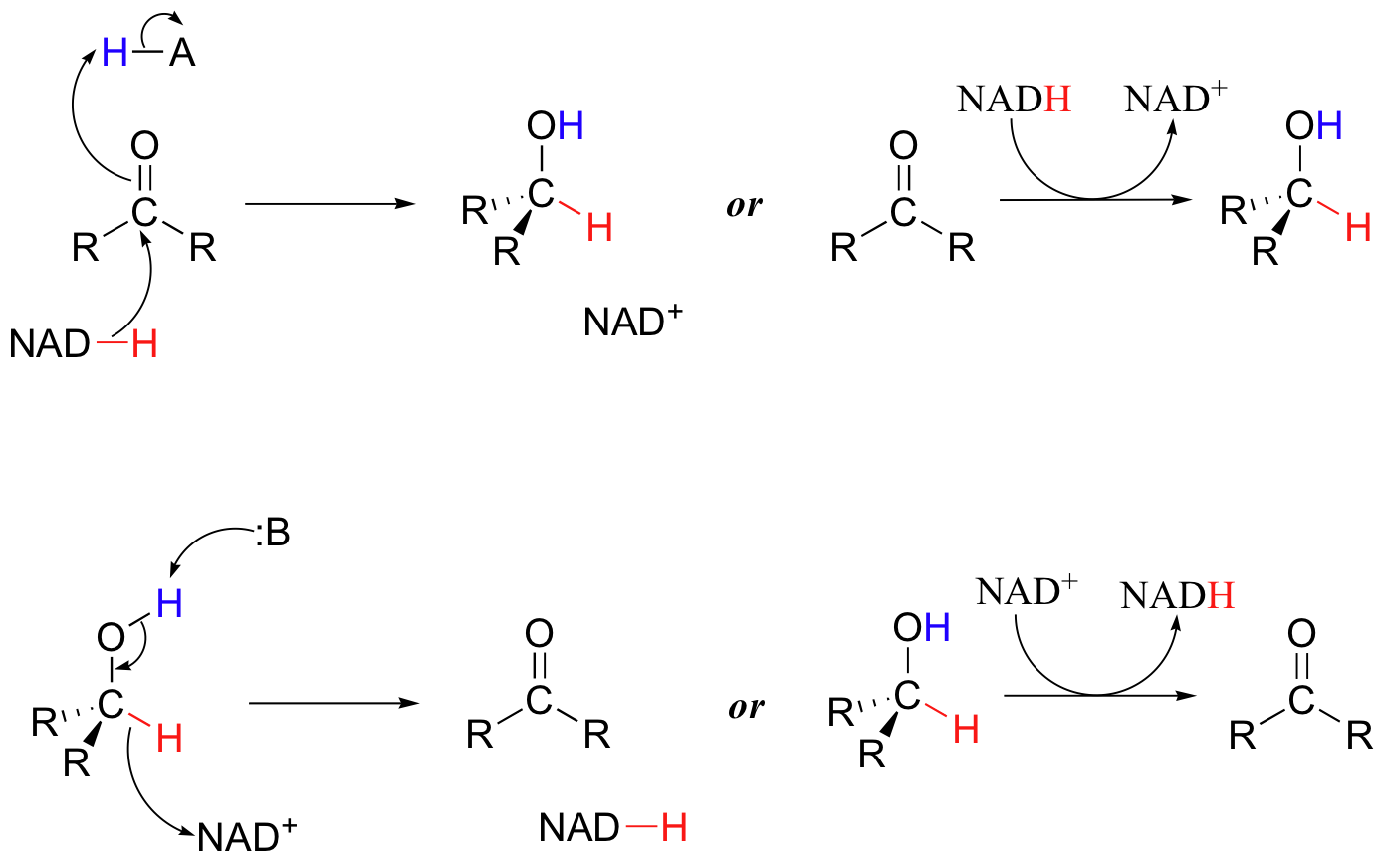
To understand how the nicotinamide cofactors function in hydride transfer, let's look at a general picture of a reversible, redox conversion from a ketone to a secondary alcohol. At the beginning of the reaction cycle, both the ketone substrate and the NADH cofactor are bound in the enzyme's active site, and C4 of the nicotinamide ring of NADH is positioned very close to the carbonyl carbon of the ketone.
As an enzymatic group transfers a proton to the ketone oxygen, the carbonyl carbon becomes more electrophilic, and is attacked by a hydride from NADH (because C4 of NADH is bound in such close proximity to the electrophile, this can occur without the generation of a free hydride ion). Notice the products of this reaction: the ketone (which accepted a hydride and a proton) has been reduced to an alcohol, and the NADH cofactor (which donated a hydride) has been oxidized to NAD+.
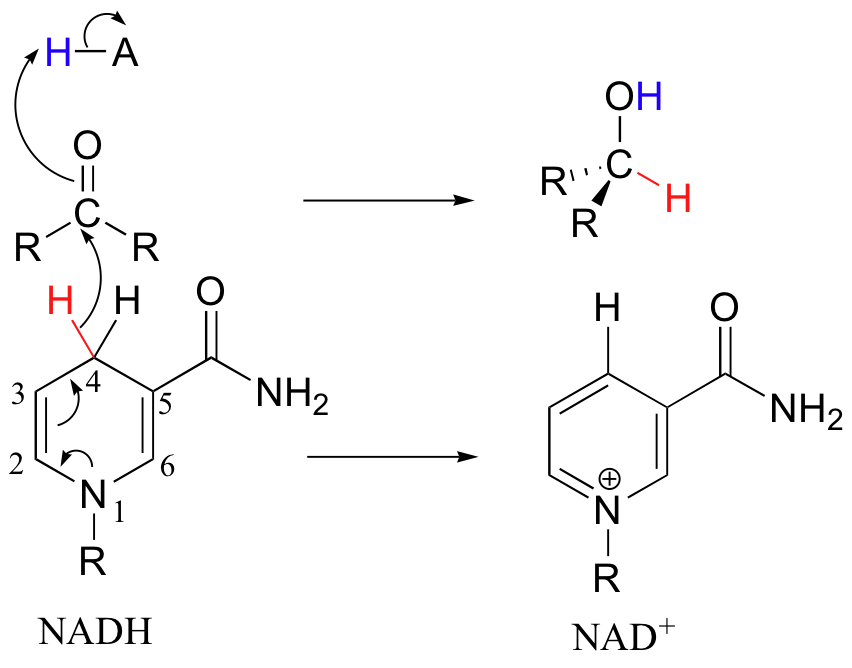
Now to the reverse reaction - the oxidation of an alcohol by NAD+.
The enzymatic base positioned above the carbonyl takes back its proton, and the electrons in the O-H bond shift down and push out the hydride, which is immediately accepted by C4 of NAD+. The products are just what we started out with - a ketone and NADH.
To simplify figures, hydrogenation and dehydrogenation reactions are often depicted in the following general ways:
16.4C: Stereochemistry of carbonyl hydrogenation and alcohol dehydrogenation
Ketone/aldehyde hydrogenase and alcohol dehydrogenase reactions are stereospecific in two distinct ways. In the hydrogenase direction, attack by the hydride can occur from either the re or the si face of an asymmetrical carbonyl, leading to the S or R alcohol.
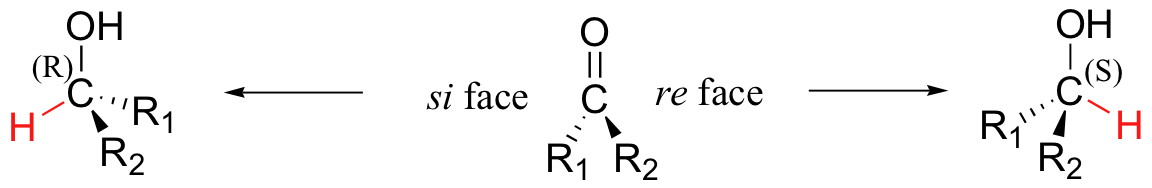
The specificity is determined by which side of the ketone or aldehyde substrate the NAD(P)H cofactor is bound in the active site.
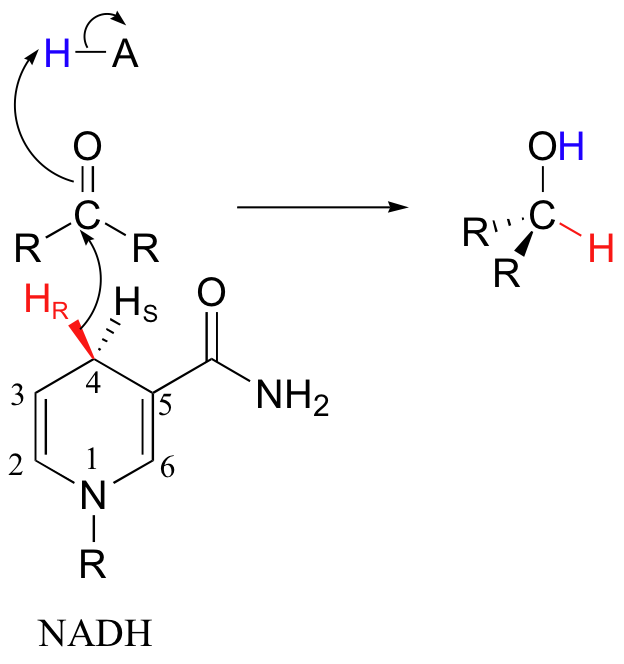
In addition, hydrogenases specifically catalyze the transfer of either the pro-R or the pro-S hydrogen at C4 of the nicotinamide ring. In the example below, the pro-R hydrogen of NADH is transferred to reduce the ketone.
The same stereochemical considerations apply in the dehydrogenase direction: in general, enzymes stereospecifically catalyze the oxidation of either an R or S alcohol, but not both (the enzyme must bind NAD(P)+ on a specific side of the substrate)
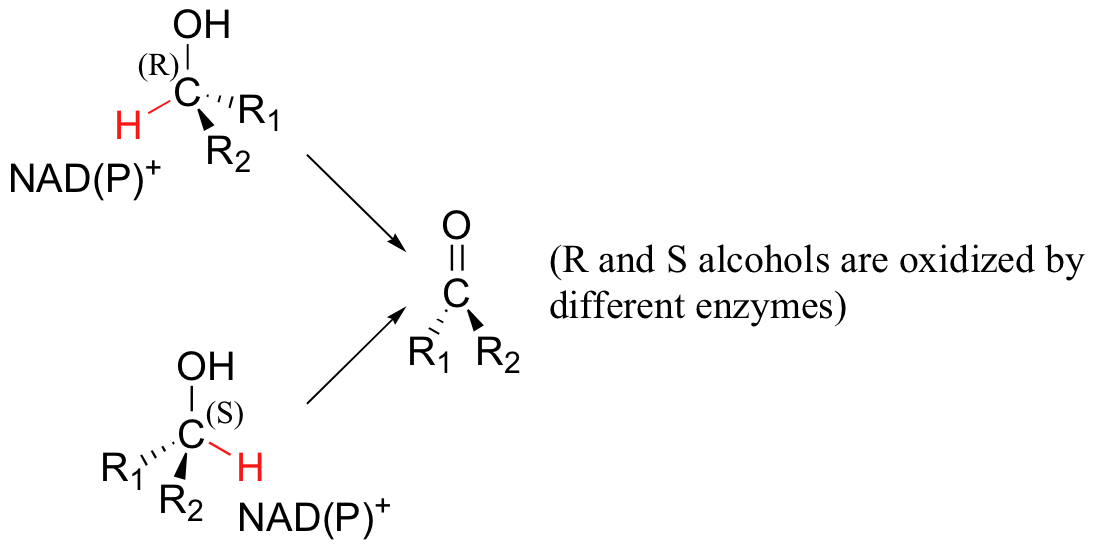
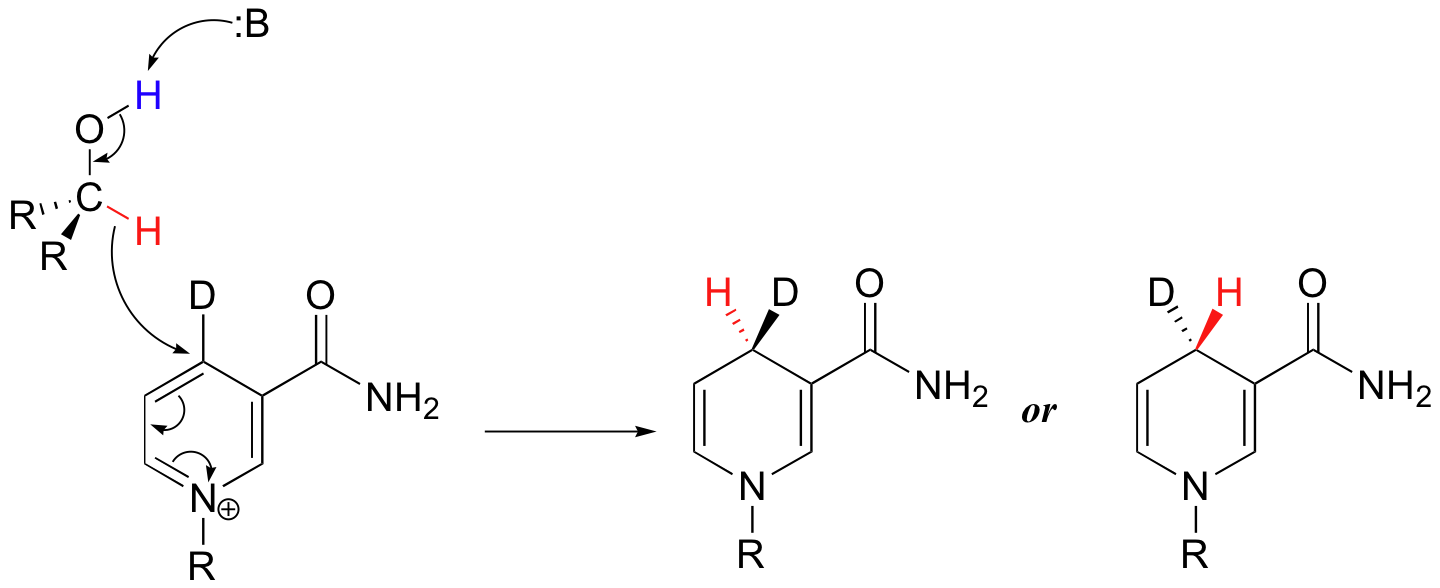
Transfer of the hydride is also specific to either the re or the si face of NAD(P)+. Dueterium labeling experiments are used to determine the stereochemistry of hydride transfers such as these.
16.4D: Examples of redox reactions involving alcohols, carbonyl groups, imines and amines
Now that we have covered the basics, let's look at some real examples of hydrogenation and dehydrogenation reactions. Although these reactions are reversible, we will show them proceeding in the direction that is most metabolically relevant.
Glycerol phosphate dehydrogenase catalyzes one of the final chemical steps in the breakdown of fat molecules. The enzyme specifically oxidizes (R)-glycerol phosphate to dihydroxyacetone phosphate, adding a hydride specifically to the re face of NAD+.
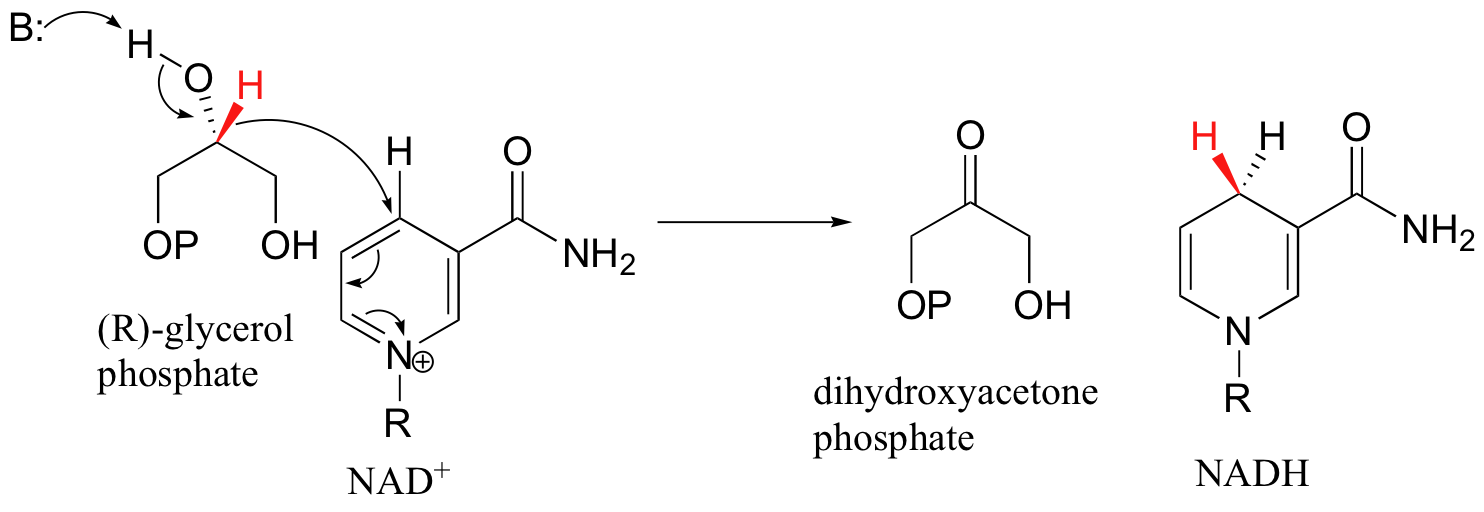
(S)-glycerol phosphate is not a substrate, and NADP+ is not an effective cofactor.
Recently, an enzyme was discovered in a class of microbes known as ‘archaea’ that catalyzes the same reaction with the opposite substrate stereochemistry (J. Biochem 1997, 122, 572). The primary metabolic role of the enzyme is apparently to catalyze the reduction of dihydroxyacetone phosphate to (S)-glycerol phosphate, with transfer of the pro-R hydrogen atom from NADPH to the re face of dihydroxyacetone phosphate..
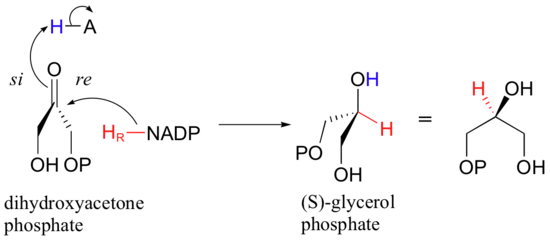
Recall (section 2.4B) that lipid molecules are constructed on a glycerol ‘backbone’. It turns out that the cell membranes of archaea are composed of lipids in which the middle carbon on the glycerol backbone has S stereochemistry, rather than the R stereochemistry that is seen in the membrane lipids of all other known forms of life. It is the enzymatic hydrogenase reaction pictured above that is responsible for this unique membrane lipid stereochemistry in archaea.
In a reaction that is very important to people in the wine and beer-making industry, an NADH-dependent enzyme in yeast produces ethanol by reducing acetaldehyde. The acetaldehyde is derived from the thiamine diphosphate-dependant decarboxylation of pyruvate that we learned about in section 14.5D.

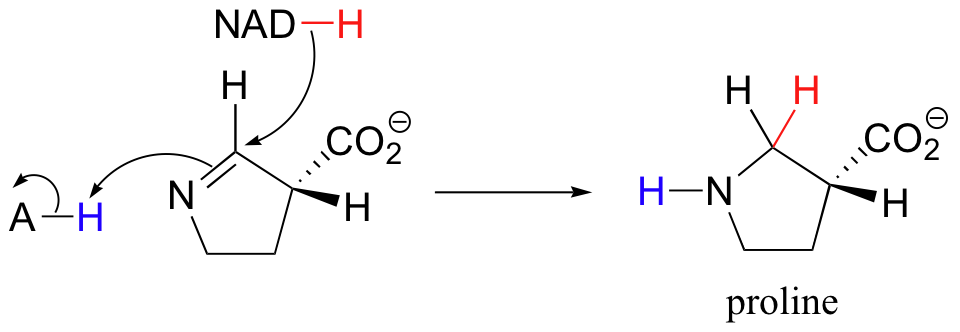
The reduction of imines to amines is mechanistically very similar to the reduction of ketones and aldehydes. The reaction below, for example, is the final step in the biosynthesis of proline.
In the dehydrogenase direction, amines can be oxidized to imines, such as in this step in the breakdown of amino acids:

The last step of this enzymatic reaction is hydrolysis of the imine to form a ketone.
The reduction of a carboxylic acid derivative to an aldehyde is essentially an acyl substitution with hydride as the nucleophile. In the example below, a thioester is reduced first to an aldehyde, then to a primary alcohol by two equivalents of NADPH. The enzyme catalyzing this reaction is the target of the statin family of cholesterol-lowering drugs.
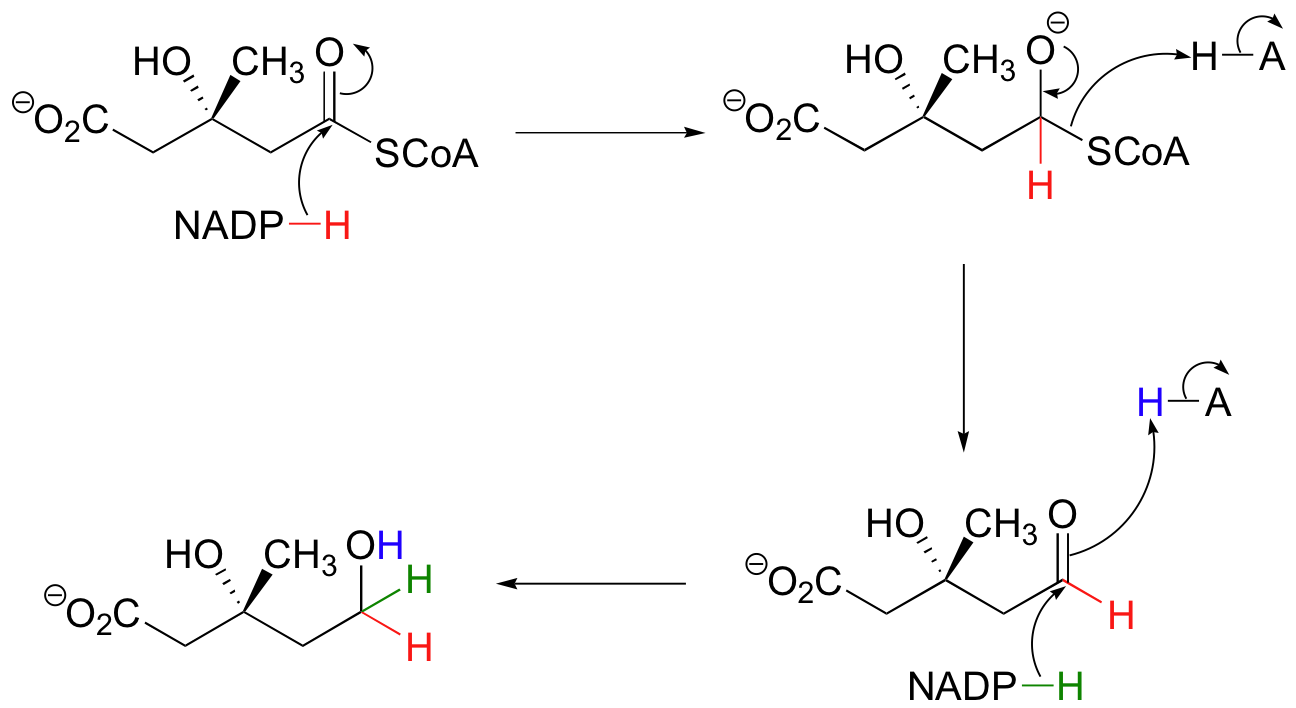
Carboxylates themselves can be reduced to aldehydes after first being converting to their activated acyl phosphate form (recall from section 12.2 that carboxylates are unreactive in acyl substitution reactions until they are activated in this way). An example is in the amino acid metabolic pathway that converts glutamate to arginine and proline.

An aldehyde is oxidized to a thioester in the first part of the reaction catalyzed by glyceraldehyde-3-phosphate dehydrogenase, a key enzyme in the glycolysis pathway.
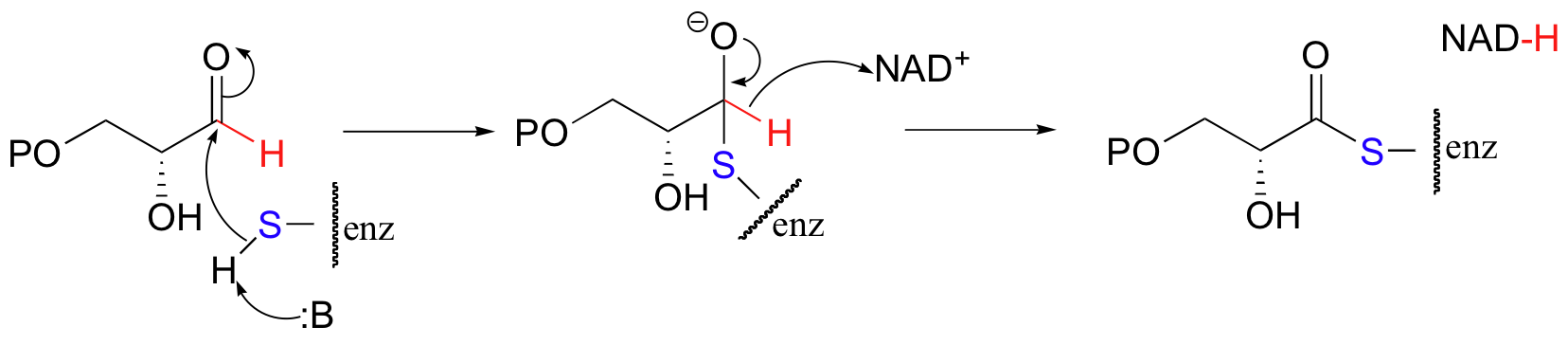
Basically this is an acyl transfer reaction with the thiol group of an active site cysteine residue as the nucleophile and a hydride as the leaving group (accepted by NAD+). The final step of the reaction (not shown) is hydrolysis of the thioester to a carboxylic acid.
The final step in the biosynthesis of the amino acid histidine provides an example of a double-oxidation from a primary alcohol to an aldehyde, then from the aldehyde to a carboxylate, with a water molecule providing the second oxygen atom.

(See Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1859 for complete details on the mechanism of this reaction.)