
6.4: Catalysis
- Page ID
- 170449

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Consider a hypothetical reaction \(\ce{R\rightarrow P}\) described by the diagram below.
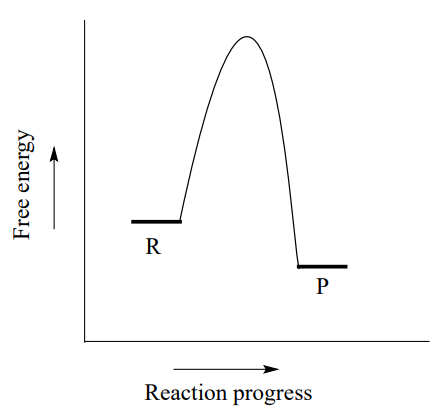
We notice two things about this reaction: it is exergonic, and it has a high activation energy. What this means is that although it is thermodynamically favorable, it is also slow: in other words, equilibrium favors product over reactants, but it will take a long time to reach equilibrium.
There are three ways that we could increase the rate of the reaction. First, we could add energy to the system by increasing the temperature, which gives the reacting molecules more energy to pass over the transition state. Increasing the temperature will increase the value of the rate constant k in the rate expression:
\[\ce{rate = k[R]}\]
In the laboratory, many organic reactions are run at high temperatures for this very purpose. We could also increase the concentration of the reaction R, which would increase the rate of the reaction without increasing the value of k.
When talking about the biochemical reactions happening in a living cell, however, increasing the reaction temperature or reactant concentration is not a reasonable option. As an alternative, we could provide a new route from point R to point P in which the activation energy is lower. The role of a catalyst is to accelerate a reaction by stabilizing the transition state, and thus lowering the activation energy.
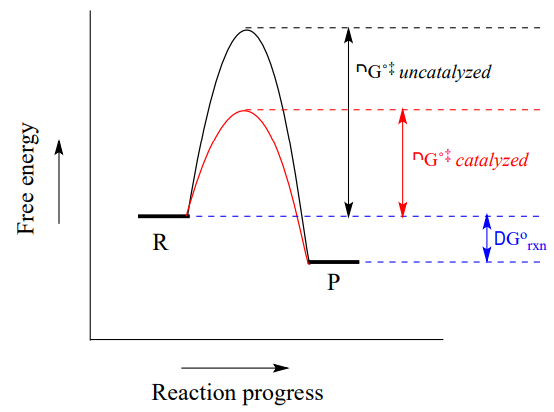
Catalysts, while they participate in the mechanism, are not consumed, so one catalyst molecule can catalyze multiple reaction cycles. Notice also that while the catalyst lowers the energy of the transition state (and thus the activation energy), it has no effect on \(\ce{\Delta G_{rxn}}\). A catalyst increases the rate of a reaction, but does not get consumed in the reaction and does not alter the equilibrium constant. In other words, a catalyst affects the kinetics of a reaction, but not the thermodynamics. Catalysts play a hugely important role in biochemical reactions.
Most organic reactions involve more than a single mechanistic step. Below is a reaction coordinate diagram illustrating rate acceleration of a two-step reaction by a catalyst:
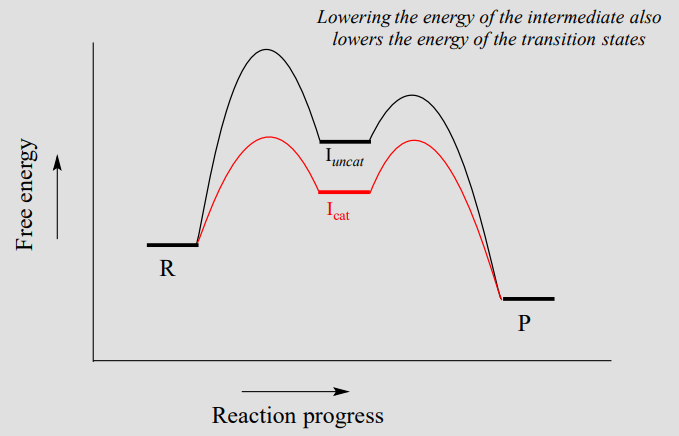
Notice that the catalyst lowers the energy of the intermediate species. A concept known as the Hammond Postulate (the details of which are beyond our scope here) tells us that when a catalyst lowers the energy of an intermediate, it also lowers the energy of the adjacent transition states. Note in this diagram above that the energy barrier for the rate-determining first step is much lower in the catalyzed reaction - thus, the overall reaction is faster. When studying how an enzyme catalyzes a biochemical reaction, chemists often are actually looking at how the enzyme interacts with - and stabilizes - an intermediate species following a rate-determining step. The Hammond postulate tells us that an understanding of enzyme-intermediate interactions will also apply to enzyme-transition state interactions.
Acids and bases as are commonly used as catalysts in organic chemistry, and chemists have come up with a huge arsenal of catalysts, many of them metals, to speed up the rates of useful laboratory reactions. Almost all biochemical reactions are catalyzed by enzymes, which are protein catalysts. In the introduction to this chapter, we heard the story of the discovery of a heat-stable DNA polymerizing enzyme which turned out to be very useful to the scientific world.
How do enzymes accomplish their role as biochemical catalysts? Recall from section 1.3 that enzymes have an active site pocket in which substrates (reactant molecules) are bound. It is inside these active site pockets that most biochemical reactions take place. Enzymes achieve catalysis in the active site by some combination of the following:
- By positioning two reacting molecules close to each other in the active site, in precisely the orientation necessary for them to react. Compare this to an uncatalyzed reaction in which completion depends on the two reactant molecules happening to collide, by chance, in the correct orientation.
- By binding substrates in such a way that they assume the proper conformation necessary for a reaction to occur.
- By increasing the reactivity of the substrates: making acidic protons more acidic, nucleophiles more nucleophilic, electrophiles more electrophilic, and leaving groups better at leaving. Very often, these feats are accomplished with acidic and/or basic amino acid side chains lining the active site pocket. As we go on to study many different types of biochemical reactions, we get a better of how this works.
- By stabilizing the transition states of the slower, rate-determining steps of the reaction. If a transition state has a negative charge, for example, the enzyme might provide a positively charged amino acid side chain, or a bound metal cation such as \(\ce{Zn^{+2}}\), as a stabilizing factor. A lower-energy transition state, of course, means a lower activation energy and a faster reaction step.
Enzymes are capable of truly amazing rate acceleration. Typical enzymes will speed up a reaction by anywhere from a million to a billion times, and the most efficient enzyme currently known to scientists is believed to accelerate its reaction by a factor of about 1017 over the uncatalyzed reaction (see Chemical and Engineering News, March 13, 2000, p. 42 for an interesting discussion about this nucleotide biosynthesis enzyme called 'orotidine monophosphate decarboxylase').
At this point, it is not necessary for you to fully understand the four ideas listed above: just keep them in mind as we go on to study a variety of common biological organic reactions and see in greater detail how enzymes have evolved to catalyze them.
Table sugar, or sucrose, is a high-energy dietary compound, as are the fats in vegetable oil. Conversion of these compounds, along with oxygen gas \(\ce{O_2}\), to water and carbon dioxide releases a lot of energy. If they are both so high in energy (in other words, thermodyamically unstable), how can they sit for years on your kitchen shelf without reacting?
Another very important point to keep in mind about enzymes is the specificity with which they catalyze reactions. We have already discussed, in chapter 3, the idea that enzymes exert a very high level of control over the stereochemistry of a reaction: if two or more stereoisomeric products could potentially form in a reaction, an enzyme will likely only catalyze the formation of one stereoisomer, with negligible formation of other side products. Likewise, enzymes demonstrate remarkable control of regiochemistry in their reactions. The glycolysis pathway enzyme glucose-6-kinase, for example, transfers a phosphate group specifically to the hydroxyl group on carbon #6 of glucose, and not to any of the other four hydroxyl groups. We'll look more closely at this reaction and others like it in chapter 9.
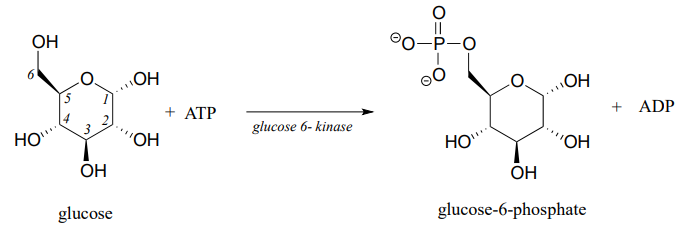
Finding ways to maintain control over stereochemistry and regiochemistry is a constant challenge for synthetic organic chemists working with non-enzymatic reactions, and the techniques that have been developed in this arena are a big part of what you will study if you go on to take a more advanced course focusing on organic synthesis.
Finally, we will encounter many biochemical reactions in this book in which the enzyme catalyzing the reaction does so with the assistance of a coenzyme. A coenzyme is a small (relative to a protein) molecule that binds in the active site of a partner enzyme and participates in some manner with the reaction being catalyzed. Table 6 at the back of the book shows the structures of several coenzymes commonly seen in biochemical reactions.